Ramuth Magalutcheemee, Sonoo Janaki, Mokshanand Fhooblall, Herring Belinda, Sofonias Tessema, McCauley John, Dinasing Ashwamed, Treurnicht Florette K
Central Health Laboratory, Victoria Hospital, Ministry of Health & Wellness, Quatre-Bornes, Plaine-Wilhems, Mauritius.
Division of Medical Virology, School of Pathology, Faculty of Health Sciences, University of Witwatersrand, Johannesburg, South Africa.
Influenza Other Respir Viruses. 2025 May;19(5):e70108. doi: 10.1111/irv.70108.
Despite being a vaccine preventable disease, influenza remains a burden in African countries. In Mauritius, influenza virus activity is year-round but evidence-based data to guide vaccination and pandemic preparedness strategies are lacking. This study aimed to describe the genetic diversity of influenza A viruses detected in Mauritius between 2017 and 2019.
Influenza A/H1N1pdm09 and A/H3N2 virus isolates were sequenced using Oxford Nanopore technology. Sequence reads assembled by CZ ID and Genome Detective web-based tools were uploaded to the EpiFlu database of the Global Initiative on Sharing All Influenza Data (GISAID). Sequence alignments and phylogenetic analysis were performed using Nextclade and MEGA XI software. BioEdit software was used to view amino acid substitutions compared to annual vaccine strains. Prediction of potential N-linked glycosylation (PNG) sites was determined by NetNGlyc 1.0.
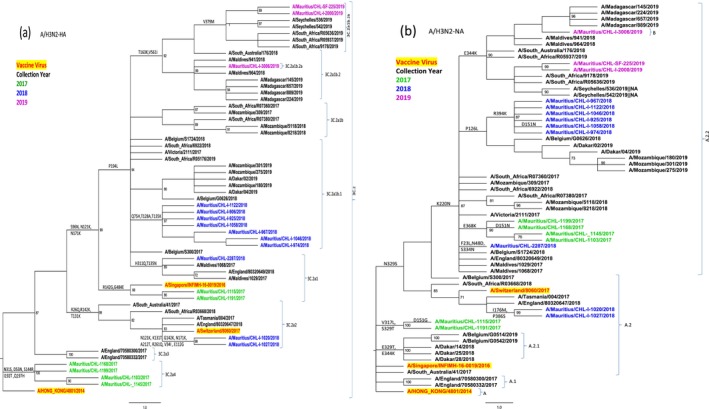
Influenza A was predominant (92.6%), with A/H1N1pdm09 prevailing overall (62.5%) but A/H3N2 dominating in 2017 (55.9%). Phylogenetic analysis identified clade 6B dominance for A/H1N1pdm09, with notable substitutions E119K, Q136K and D151E linked to antigenic changes. A/H3N2 exhibited significant genetic diversity, with co-circulation of 3C.2a4 and 3C.2a1 in 2017 while 2018 predominant subclade 3C.2a1b.1 highlights continued antigenic drift. Loss of PNG sites at position 158 (11/21; 52.4%) in HA and position 329 (81.0%, 17/21) in NA of A/H3N2 viruses were observed.
Continued evolution of A/H1N1pdm09 and A/H3N2 viruses in Mauritius highlights the need for sustained genomic surveillance to inform vaccine and antiviral strategies. Data from Mauritius will contribute to understanding of influenza viruses' ecology in the African region and globally.
尽管流感是一种可通过疫苗预防的疾病,但在非洲国家仍然是一个负担。在毛里求斯,流感病毒全年都有活动,但缺乏指导疫苗接种和大流行防范策略的循证数据。本研究旨在描述2017年至2019年在毛里求斯检测到的甲型流感病毒的基因多样性。
使用牛津纳米孔技术对甲型H1N1pdm09和甲型H3N2病毒分离株进行测序。由CZ ID和基于网络的Genome Detective工具组装的序列读数被上传到全球共享流感数据倡议组织(GISAID)的EpiFlu数据库。使用Nextclade和MEGA XI软件进行序列比对和系统发育分析。使用BioEdit软件查看与年度疫苗株相比的氨基酸替换。通过NetNGlyc 1.0确定潜在N-连接糖基化(PNG)位点的预测。
甲型流感占主导地位(92.6%),总体上甲型H1N1pdm09占优势(62.5%),但甲型H3N2在2017年占主导(55.9%)。系统发育分析确定甲型H1N1pdm09的6B分支占主导地位,与抗原性变化相关的显著替换E119K、Q136K和D151E。甲型H3N2表现出显著的基因多样性,2017年3C.2a4和3C.2a1共同流行,而2018年占主导的亚分支3C.2a1b.1突出了持续的抗原漂移。在甲型H3N2病毒的HA的158位(11/21;52.4%)和NA的329位(81.0%,17/21)观察到PNG位点的缺失。
毛里求斯甲型H1N1pdm09和甲型H3N2病毒的持续进化凸显了持续进行基因组监测以指导疫苗和抗病毒策略的必要性。来自毛里求斯的数据将有助于了解非洲地区和全球流感病毒的生态学。