Volloch Vladimir, Rits-Volloch Sophia
Department of Developmental Biology, Harvard School of Dental Medicine, Boston, MA 02115, USA.
Division of Molecular Medicine, Children's Hospital, Boston, MA 02115, USA.
Int J Mol Sci. 2025 Apr 29;26(9):4252. doi: 10.3390/ijms26094252.
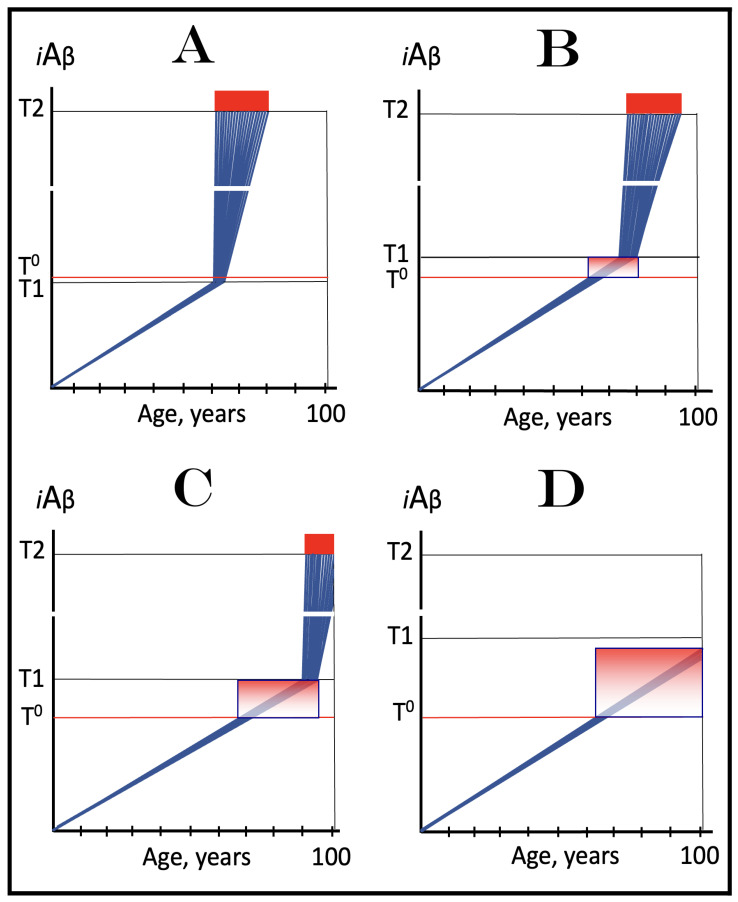
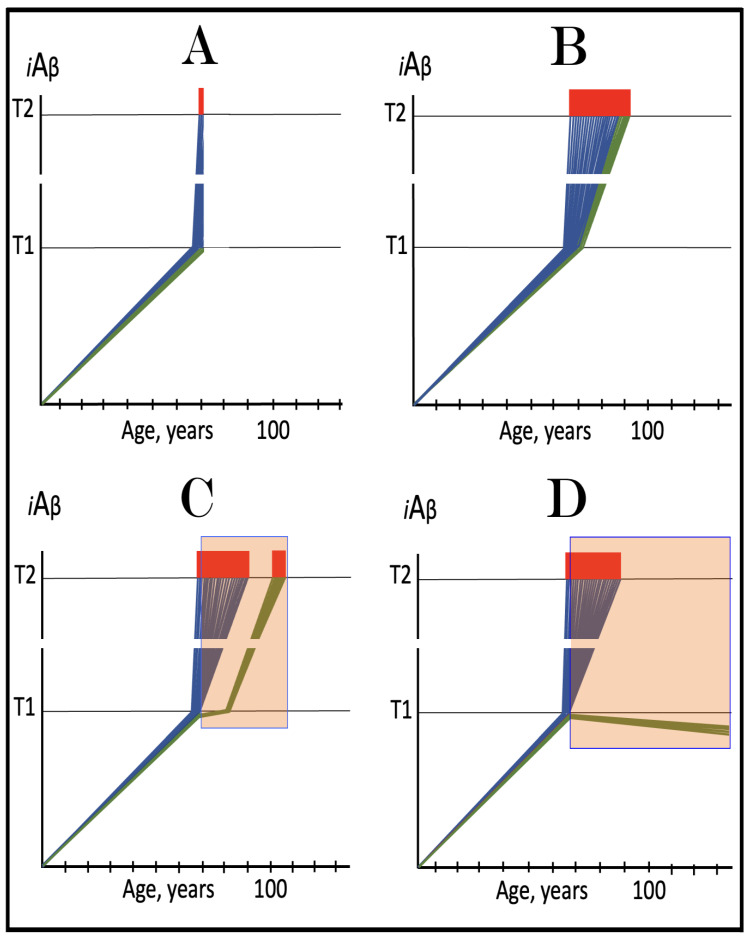
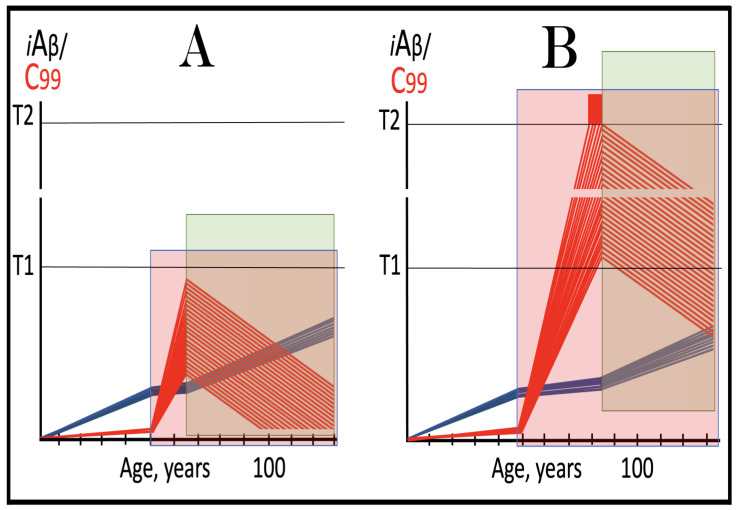
The present analyzes the remarkable evolution of the Amyloid Cascade Hypothesis 2.0 (ACH2.0) theory of Alzheimer's disease (AD) since its inception a few years ago, as reflected in the diminishing role of amyloid-beta (Aβ) in the disease. In the initial iteration of the ACH2.0, Aβ-protein-precursor (AβPP)-derived intraneuronal Aβ (iAβ), accumulated to neuronal integrated stress response (ISR)-eliciting levels, triggers AD. The neuronal ISR, in turn, activates the AβPP-independent production of its C99 fragment that is processed into iAβ, which drives the disease. The second iteration of the ACH2.0 stemmed from the realization that AD is, in fact, a disease of the sustained neuronal ISR. It introduced two categories of AD-conventional and unconventional-differing mainly in the manner of their causation. The former is caused by the neuronal ISR triggered by AβPP-derived iAβ, whereas in the latter, the neuronal ISR is elicited by stressors distinct from AβPP-derived iAβ and arising from brain trauma, viral and bacterial infections, and various types of inflammation. Moreover, conventional AD always contains an unconventional component, and in both forms, the disease is driven by iAβ generated independently of AβPP. In its third, the current, iteration, the ACH2.0 posits that proteolytic production of Aβ is suppressed in AD-affected neurons and that the disease is driven by C99 generated independently of AβPP. Suppression of Aβ production in AD seems an oxymoron: Aβ is equated with AD, and the later is inconceivable without the former in an ingrained Amyloid Cascade Hypothesis (ACH)-based notion. But suppression of Aβ production in AD-affected neurons is where the logic leads, and to follow it we only need to overcome the inertia of the preexisting assumptions. Moreover, not only is the generation of Aβ suppressed, so is the production of all components of the AβPP proteolytic pathway. This assertion is not a quantum leap (unless overcoming the inertia counts as such): the global cellular protein synthesis is severely suppressed under the neuronal ISR conditions, and there is no reason for constituents of the AβPP proteolytic pathway to be exempted, and they, apparently, are not, as indicated by the empirical data. In contrast, tau protein translation persists in AD-affected neurons under ISR conditions because the human tau mRNA contains an internal ribosomal entry site in its 5'UTR. In current mouse models, iAβ derived from AβPP expressed exogenously from human transgenes elicits the neuronal ISR and thus suppresses its own production. Its levels cannot principally reach AD pathology-causing levels regardless of the number of transgenes or the types of FAD mutations that they (or additional transgenes) carry. Since the AβPP-independent C99 production pathway is inoperative in mice, the current transgenic models have no potential for developing the full spectrum of AD pathology. What they display are only effects of the AβPP-derived iAβ-elicited neuronal ISR. The paper describes strategies to construct adequate transgenic AD models. It also details the utilization of human neuronal cells as the only adequate model system currently available for conventional and unconventional AD. The final alteration of the ACH2.0, introduced in the present , is that AβPP, which supports neuronal functionality and viability, is, after all, potentially produced in AD-affected neurons, albeit not conventionally but in an ISR-driven and -compatible process. Thus, the present narrative begins with the "omnipotent" Aβ capable of both triggering and driving the disease and ends up with this peptide largely dislodged from its pedestal and retaining its central role in triggering the disease in only one, although prevalent (conventional), category of AD (and driving it in none). Among interesting inferences of the present is the determination that "sporadic AD" is not sporadic at all ("non-familial" would be a much better designation). The term has fatalistic connotations, implying that the disease can strike at random. This is patently not the case: The conventional disease affects a distinct subpopulation, and the basis for unconventional AD is well understood. Another conclusion is that, unless prevented, the occurrence of conventional AD is inevitable given a sufficiently long lifespan. This also defines therapeutic directions not to be taken as well as auspicious ways forward. The former category includes ACH-based drugs (those interfering with the proteolytic production of Aβ and/or depleting extracellular Aβ). They are legitimate (albeit inefficient) preventive agents for conventional AD. There is, however, a proverbial snowball's chance in hell of them being effective in symptomatic AD, lecanemab, donanemab, and any other "…mab" or "…stat" notwithstanding. They comprise Aβ-specific antibodies, inhibitors of beta- and gamma-secretase, and modulators of the latter. In the latter category, among ways to go are the following: (1) Depletion of iAβ, which, if sufficiently "deep", opens up a tantalizing possibility of once-in-a-lifetime preventive transient treatment for conventional AD and aging-associated cognitive decline, AACD. (2) Composite therapy comprising the degradation of C99/iAβ and concurrent inhibition of the neuronal ISR. A single transient treatment could be sufficient to arrest the progression of conventional AD and prevent its recurrence for life. Multiple recurrent treatments would achieve the same outcome in unconventional AD. Alternatively, the sustained reduction/removal of unconventional neuronal ISR-eliciting stressors through the elimination of their source would convert unconventional AD into conventional one, preventable/treatable by a single transient administration of the composite C99/iAβ depletion/ISR suppression therapy. Efficient and suitable ISR inhibitors are available, and it is explicitly clear where to look for C99/iAβ-specific targeted degradation agents-activators of BACE1 and, especially, BACE2. Directly acting C99/iAβ-specific degradation agents such as proteolysis-targeting chimeras (PROTACs) and molecular-glue degraders (MGDs) are also viable options. (3) A circumscribed shift (either upstream or downstream) of the position of transcription start site (TSS) of the human AβPP gene, or, alternatively, a gene editing-mediated excision or replacement of a small, defined segment of its portion encoding 5'-untranslated region of AβPP mRNA; targeting AβPP RNA with anti-antisense oligonucleotides is another possibility. If properly executed, these RNA-based strategies would not interfere with the protein-coding potential of AβPP mRNA, and each would be capable of both preventing and stopping the AβPP-independent generation of C99 and thus of either preventing AD or arresting the progression of the disease in its conventional and unconventional forms. The paper is interspersed with "validation" sections: every conceptually significant notion is either validated by the existing data or an experimental procedure validating it is proposed.
本文分析了自几年前提出以来,阿尔茨海默病(AD)的淀粉样蛋白级联假说2.0(ACH2.0)理论的显著演变,这体现在β淀粉样蛋白(Aβ)在该疾病中的作用逐渐减弱。在ACH2.0的最初版本中,由淀粉样前体蛋白(AβPP)衍生的神经元内Aβ(iAβ)积累到引发神经元综合应激反应(ISR)的水平,从而引发AD。反过来,神经元ISR会激活其C99片段的AβPP非依赖性产生,该片段会被加工成iAβ,进而推动疾病发展。ACH2.0的第二个版本源于这样的认识,即AD实际上是一种持续性神经元ISR的疾病。它引入了两类AD——传统型和非传统型,主要区别在于其病因。前者由AβPP衍生的iAβ引发的神经元ISR引起,而在后者中,神经元ISR由与AβPP衍生的iAβ不同的应激源引发,这些应激源来自脑外伤、病毒和细菌感染以及各种类型的炎症。此外,传统AD总是包含一个非传统成分,并且在这两种形式中,疾病都是由独立于AβPP产生的iAβ驱动的。在其第三个版本,即当前版本中,ACH2.0假定在受AD影响的神经元中Aβ的蛋白水解产生受到抑制,并且疾病是由独立于AβPP产生的C99驱动的。在AD中抑制Aβ产生似乎是一种矛盾说法:Aβ与AD等同,并且在基于根深蒂固的淀粉样蛋白级联假说(ACH)的观念中,没有前者后者是不可想象的。但逻辑推导的结果是,在受AD影响的神经元中Aβ产生受到抑制,要遵循这一观点我们只需克服现有假设的惯性。此外,不仅Aβ的产生受到抑制,AβPP蛋白水解途径的所有成分的产生也受到抑制。这一断言并非巨大飞跃(除非克服惯性也算):在神经元ISR条件下,全局细胞蛋白质合成受到严重抑制;AβPP蛋白水解途径的成分没有理由豁免,而经验数据表明它们显然也没有豁免。相比之下,在ISR条件下,tau蛋白翻译在受AD影响的神经元中持续存在,因为人类tau mRNA在其5'UTR中包含一个内部核糖体进入位点。在当前的小鼠模型中,源自人类转基因外源表达的AβPP的iAβ引发神经元ISR,从而抑制其自身产生。无论转基因数量或它们(或其他转基因)携带的早发性阿尔茨海默病(FAD)突变类型如何,其水平原则上都无法达到导致AD病理的水平。由于AβPP非依赖性C99产生途径在小鼠中不起作用,当前的转基因模型没有发展出完整AD病理谱的潜力。它们所显示的只是AβPP衍生的iAβ引发的神经元ISR的影响。本文描述了构建合适转基因AD模型的策略。它还详细介绍了将人类神经元细胞作为目前唯一适用于传统型和非传统型AD的模型系统的应用。本文引入的ACH2.0的最终改变是,支持神经元功能和活力且原本可能在受AD影响的神经元中产生的AβPP,尽管不是以传统方式,而是在一个由ISR驱动且与之兼容的过程中产生。因此,本文叙述从能够引发和驱动疾病的“全能”Aβ开始,最终以这种肽在很大程度上从其主导地位被取代结束,并且它仅在一种(尽管普遍存在的传统型)AD类别中在引发疾病方面保留其核心作用(在驱动疾病方面则没有)。本文有趣的推断之一是确定“散发性AD”根本不是散发性的(“非家族性”会是一个更好的称呼)。这个术语带有宿命论的含义,暗示疾病可能随机发作。显然并非如此:传统型疾病影响一个独特的亚群,并且非传统型AD的病因已得到充分理解。另一个结论是,除非得到预防,给定足够长的寿命,传统型AD的发生是不可避免的。本文还定义了不应采取的治疗方向以及有利的前进方向。前一类包括基于ACH的药物(那些干扰Aβ的蛋白水解产生和/或消耗细胞外Aβ的药物)。它们是传统型AD的合理(尽管低效)预防剂。然而,尽管有lecanemab、donanemab以及任何其他“……mab ”或“……stat ”,它们在症状性AD中有效的可能性微乎其微。它们包括Aβ特异性抗体、β和γ分泌酶抑制剂以及后者的调节剂。在后一类中,可行的方法包括以下几种:(1)消耗iAβ,如果消耗得足够“彻底”,就为传统型AD和与衰老相关的认知衰退(AACD)开辟了一种一生仅一次的预防性短暂治疗的诱人可能性。(2)包括C99/iAβ降解和同时抑制神经元ISR的联合疗法。单次短暂治疗可能足以阻止传统型AD的进展并预防其终生复发。多次重复治疗在非传统型AD中会取得相同的结果。或者,通过消除其来源持续减少/消除引发非传统型神经元ISR的应激源,将非传统型AD转化为传统型AD,通过单次短暂给予C99/iAβ消耗/ISR抑制联合疗法即可预防/治疗。有高效且合适的ISR抑制剂,并且明确知道在哪里可以找到C99/iAβ特异性靶向降解剂——β分泌酶1(BACE1)的激活剂,尤其是BACE2的激活剂。直接作用的C99/iAβ特异性降解剂,如蛋白酶靶向嵌合体(PROTACs)和分子胶降解剂(MGDs)也是可行的选择。(3)人类AβPP基因转录起始位点(TSS)位置的有限移动(向上游或下游),或者通过基因编辑介导切除或替换其编码AβPP mRNA 5'非翻译区的一小段特定片段;用反义寡核苷酸靶向AβPP RNA是另一种可能性。如果执行得当,这些基于RNA的策略不会干扰AβPP mRNA的蛋白质编码潜力,并且每种策略都能够预防和阻止AβPP非依赖性C99的产生,从而预防AD或以传统和非传统形式阻止疾病进展。本文穿插了“验证”部分:每个具有概念重要性的概念要么由现有数据验证,要么提出了验证它的实验程序。