Dyroff Antonia I, López-Valiñas Álvaro, Magalhaes Humberto B, Podico Giorgia, Canisso Igor F, Almiñana Carmen, Bauersachs Stefan
Institute of Veterinary Anatomy, Vetsuisse Faculty Zurich, University of Zurich, Lindau (ZH), Switzerland.
College of Veterinary Medicine, University of Illinois Urbana-Champaign, Urbana, IL, USA.
Sci Rep. 2025 May 16;15(1):17037. doi: 10.1038/s41598-025-00969-5.
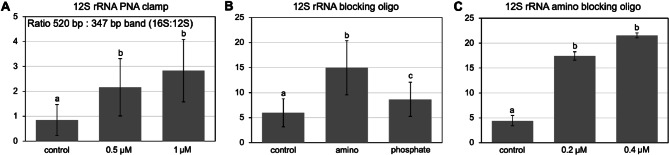
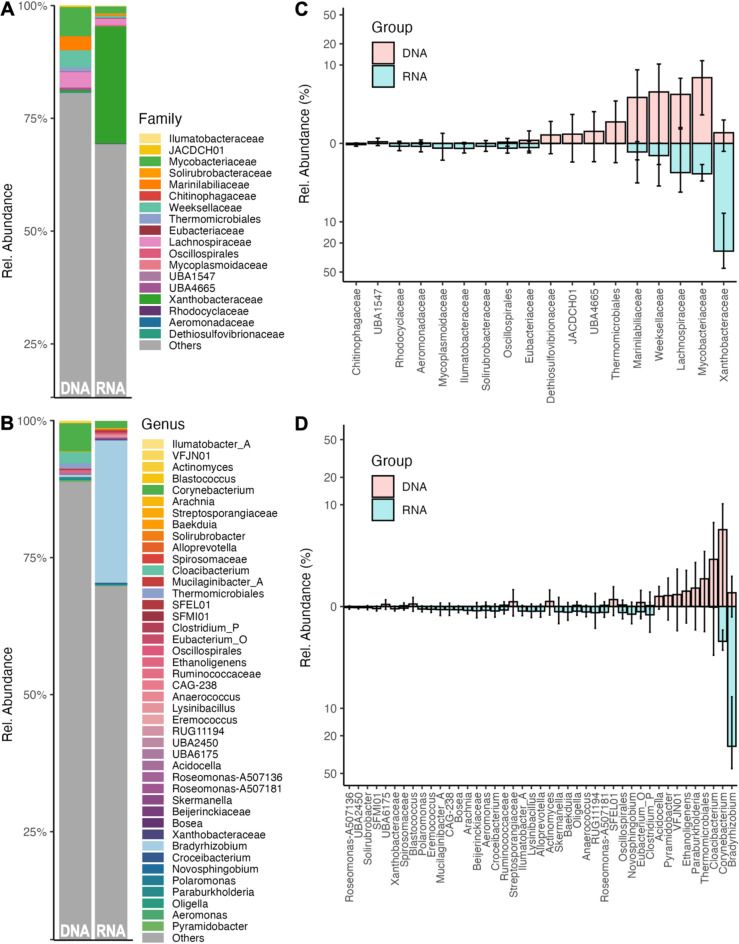
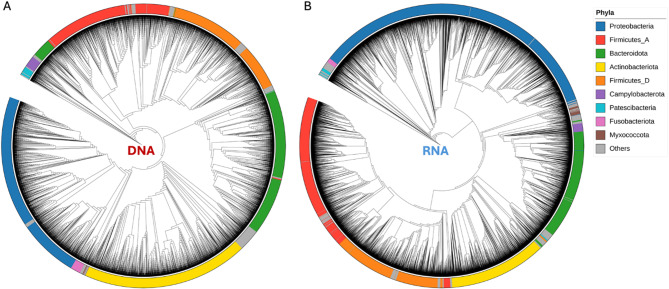
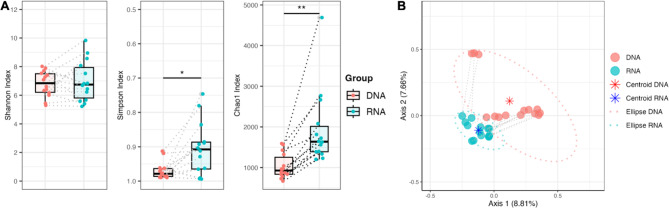
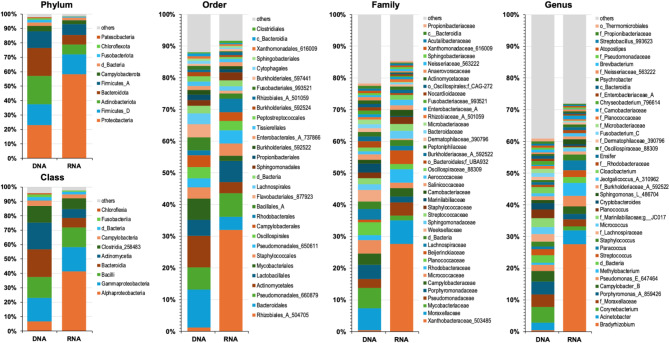
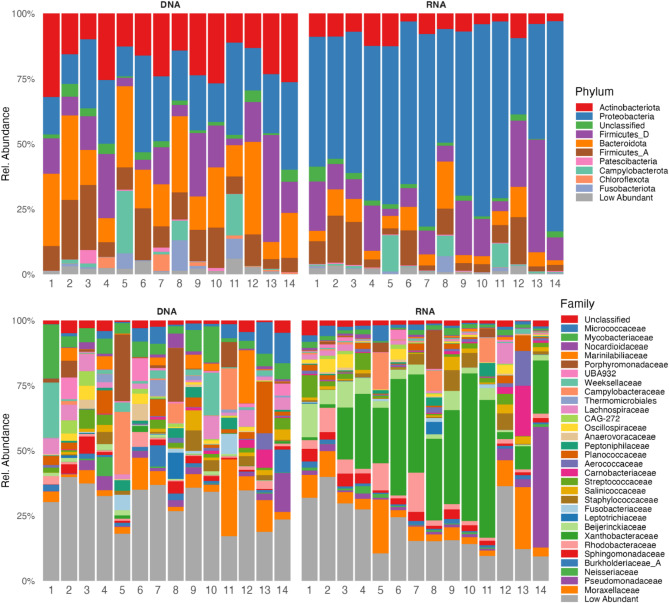
Studies in humans and large animals indicate a relationship between the uterine microbiome composition and endometrial receptivity. Despite many studies have been performed, the analysis of the uterine microbiome remains challenging due to the very low microbial biomass. Studies in other biological systems showed that RNA-based microbiome analysis complements DNA-based results and provides information about active bacteria in a sample. Thus, the aim of this study was to establish a highly sensitive and specific 16S rRNA gene V3-V4 amplicon PCR from equine uterine cytobrush samples and to compare DNA- and RNA-based 16S rRNA microbiome analysis. An optimized 16S rRNA gene V3-V4 amplicon PCR protocol from equine uterine cytobrush samples was developed, which was able to detect less than 38 bacterial genome copies using a bacterial DNA community standard. For the RNA-based amplicon generation protocol starting from cDNA, at least a 10-fold higher sensitivity was estimated compared to DNA-based approach. The comparison of using RNA and DNA isolated from the same uterine cytobrush samples as input for 16S V3-V4 amplicon sequencing revealed a much higher number of amplicon sequence variants as well as taxonomic units for the RNA-based approach. This resulted in significant differences in alpha (Simpson, Chao1) and beta diversity between RNA- and DNA-based analysis. Differential abundance analysis revealed significant differences between DNA and RNA samples at all taxonomic levels. Despite these differences, the overall microbiome composition was similar between the paired DNA and RNA samples. Many differences were probably found due to the higher sensitivity of the RNA-based approach. Furthermore, the DNA-based analysis is biased by the rRNA gene copy numbers (1-21), and the RNA-based analysis by the number of ribosomes per cell, which was reflected in the differences in the microbiome composition between the approaches. In addition, the results suggested that the DNA-based analysis is detecting cell-free bacterial DNA and/or DNA of dead bacteria that could be present in the samples. Altogether, the obtained results indicate advantages of a combined DNA- and RNA-based microbiome analysis, offering complementary and valuable information in the context of fertility-related studies of the uterine microbiome.
对人类和大型动物的研究表明,子宫微生物群组成与子宫内膜容受性之间存在关联。尽管已经进行了许多研究,但由于微生物生物量极低,子宫微生物群的分析仍然具有挑战性。在其他生物系统中的研究表明,基于RNA的微生物群分析可以补充基于DNA的结果,并提供有关样本中活性细菌的信息。因此,本研究的目的是从马子宫细胞刷样本中建立一种高度灵敏且特异的16S rRNA基因V3-V4扩增子PCR,并比较基于DNA和RNA的16S rRNA微生物群分析。开发了一种优化的来自马子宫细胞刷样本的16S rRNA基因V3-V4扩增子PCR方案,该方案使用细菌DNA群落标准能够检测到少于38个细菌基因组拷贝。对于从cDNA开始的基于RNA的扩增子生成方案,与基于DNA的方法相比,估计灵敏度至少高10倍。将从相同子宫细胞刷样本中分离的RNA和DNA用作16S V3-V4扩增子测序的输入进行比较,结果显示基于RNA的方法具有更多的扩增子序列变体以及分类单元。这导致基于RNA和DNA的分析在α(辛普森指数、Chao1指数)和β多样性方面存在显著差异。差异丰度分析显示,在所有分类水平上,DNA和RNA样本之间存在显著差异。尽管存在这些差异,但配对的DNA和RNA样本之间的总体微生物群组成相似。可能由于基于RNA的方法具有更高的灵敏度而发现了许多差异。此外,基于DNA的分析受rRNA基因拷贝数(1-21)的影响,而基于RNA的分析受每个细胞核糖体数量的影响,这反映在两种方法之间微生物群组成的差异上。此外,结果表明基于DNA的分析检测的是样本中可能存在的无细胞细菌DNA和/或死细菌的DNA。总之,获得的结果表明基于DNA和RNA的微生物群联合分析具有优势,在子宫微生物群与生育相关的研究中提供了互补且有价值的信息。