Venkatesan Arunkumar, Bernstein Audrey M
Department of Ophthalmology and Visual Sciences, SUNY Upstate Medical University, Syracuse, NY, United States.
New York VA Healthcare, Syracuse VA Medical Center, Syracuse, NY, United States.
Front Cell Dev Biol. 2025 Apr 25;13:1595121. doi: 10.3389/fcell.2025.1595121. eCollection 2025.
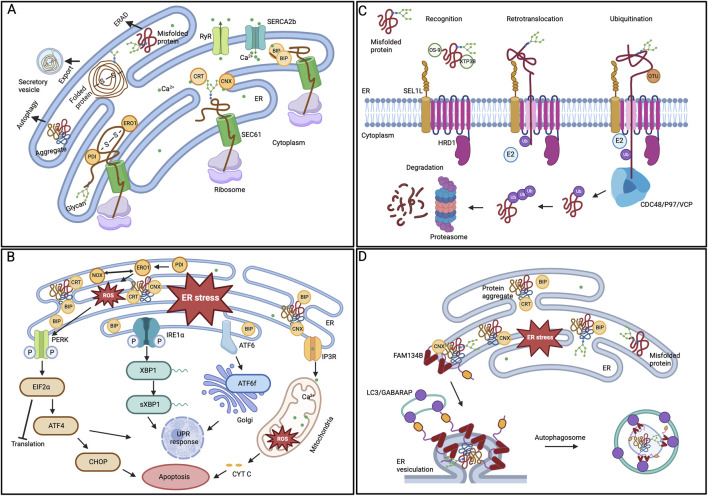
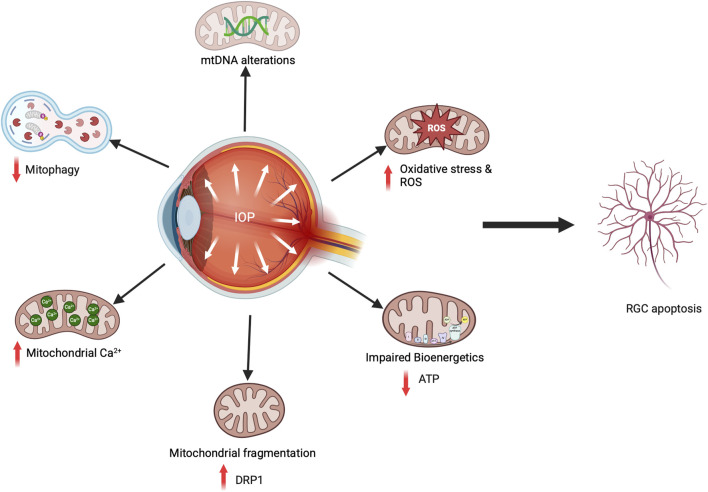
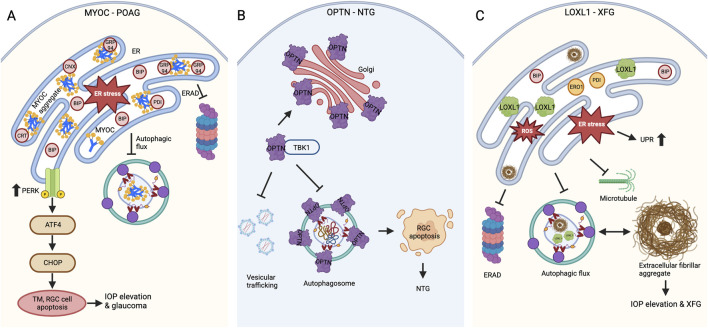
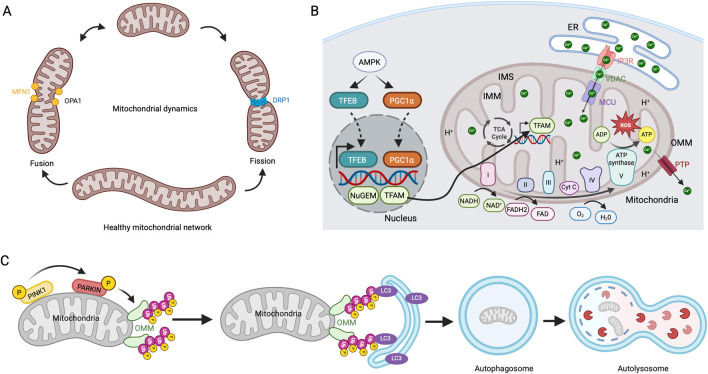
Glaucoma is a leading cause of irreversible blindness worldwide. Elevated intraocular pressure caused by restricted outflow of the aqueous humor leads to the degeneration of retinal ganglion cells (RGCs) and their axons. Emerging evidence suggests that pathological mechanisms relating to protein folding and mitochondrial dysfunction are significant factors in the disease onset of different types of open-angle glaucoma. In this review, we discuss these defects in three distinct types of open-angle glaucoma: primary open-angle glaucoma (POAG), normal tension glaucoma (NTG), and exfoliation glaucoma (XFG). Genetic mutations linked to the previously mentioned open-angle glaucoma, including those in myocilin (MYOC), optineurin (OPTN), and lysyl oxidase 1 (LOXL1), disrupt protein folding and homeostasis, leading to endoplasmic reticulum stress, activation of the unfolded protein response and impaired autophagic protein degradation. These factors contribute to trabecular meshwork and retinal ganglion cell apoptosis. In addition to protein folding defects, mitochondrial dysfunction is also associated with the progression of trabecular meshwork damage and the death of RGCs. Factors such as oxidative stress, an altered mitochondrial fission-fusion balance, and mitophagy dysregulation make RGCs vulnerable and contribute to optic nerve degeneration. The crosstalk between protein folding and mitochondrial defects in glaucoma underscores the complexity of disease pathogenesis and offers potential targets for therapeutic intervention. Strategies aimed at restoring protein homeostasis, enhancing mitochondrial function, and mitigating cellular stress responses hold promise for neuroprotection in glaucoma.
青光眼是全球不可逆性失明的主要原因。房水流出受限导致眼内压升高,进而引起视网膜神经节细胞(RGCs)及其轴突的退化。新出现的证据表明,与蛋白质折叠和线粒体功能障碍相关的病理机制是不同类型开角型青光眼发病的重要因素。在这篇综述中,我们讨论三种不同类型开角型青光眼的这些缺陷:原发性开角型青光眼(POAG)、正常眼压性青光眼(NTG)和剥脱性青光眼(XFG)。与上述开角型青光眼相关的基因突变,包括肌纤蛋白(MYOC)、视紫质(OPTN)和赖氨酰氧化酶1(LOXL1)中的突变,会破坏蛋白质折叠和内环境稳定,导致内质网应激、未折叠蛋白反应的激活以及自噬性蛋白质降解受损。这些因素导致小梁网和视网膜神经节细胞凋亡。除了蛋白质折叠缺陷外,线粒体功能障碍也与小梁网损伤的进展和RGCs的死亡有关。氧化应激、线粒体裂变 - 融合平衡改变和线粒体自噬失调等因素使RGCs变得脆弱,并导致视神经变性。青光眼患者蛋白质折叠与线粒体缺陷之间的相互作用突出了疾病发病机制的复杂性,并为治疗干预提供了潜在靶点。旨在恢复蛋白质稳态、增强线粒体功能和减轻细胞应激反应的策略有望为青光眼的神经保护提供帮助。