Wang Han, Bing Ziyan, Li Lu, Gao Ziwen, Nwanade Chuks Fidelis, Dong Na, Li Ke, Du Leyan, Yu Zhijun
Hebei Key Laboratory of Animal Physiology, Biochemistry and Molecular Biology, Hebei Collaborative Innovation Center for Eco-Environment, Ministry of Education Key Laboratory of Molecular and Cellular Biology, College of Life Sciences, Hebei Normal University, Shijiazhuang, 050024, China.
Guangdong Key Laboratory of Animal Conservation and Resource Utilization, Guangdong Public Laboratory of Wild Animal Conservation and Utilization, Institute of Zoology, Guangdong Academy of Sciences, Guangzhou, 510260, China.
Parasit Vectors. 2025 Jun 1;18(1):202. doi: 10.1186/s13071-025-06810-2.
Haemaphysalis longicornis is an important vector that transmits a variety of pathogens to humans and animals. This tick species is unique for having two separate reproductive populations: bisexual and parthenogenetic populations. In bisexual populations, morphological differences exist between the males and females, with the females often larger than the males. DNA methylation, as a key epigenetic modification, plays a crucial role in biological processes such as the maintenance of normal cellular function, the regulation of gene expression, and embryonic development. However, the epigenetic mechanism underlying sex differentiation in the bisexual population of H. longicornis has been overlooked.
In the present study, the global DNA methylation profiles of the female and male H. longicornis ticks from the bisexual population were explored using whole-genome bisulfite sequencing. Differentially methylated regions (DMRs) were identified, followed by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of DMR-related genes.
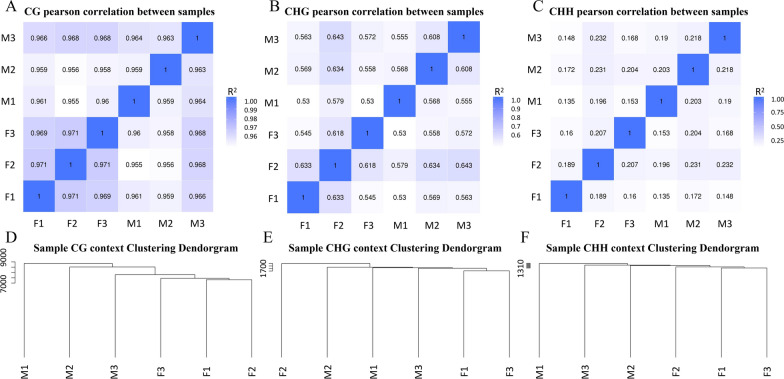
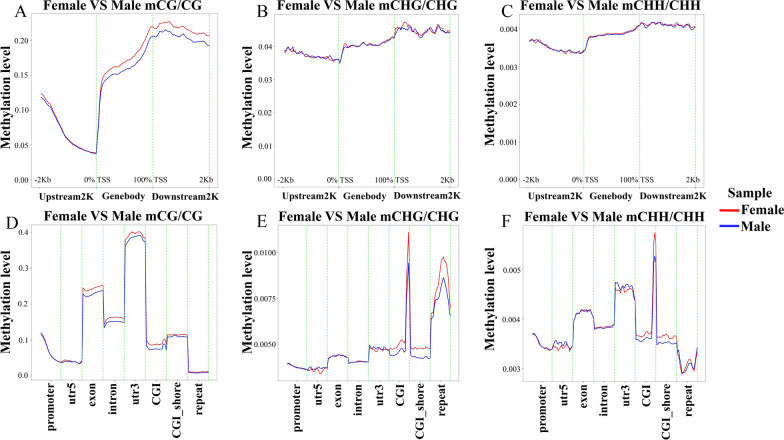
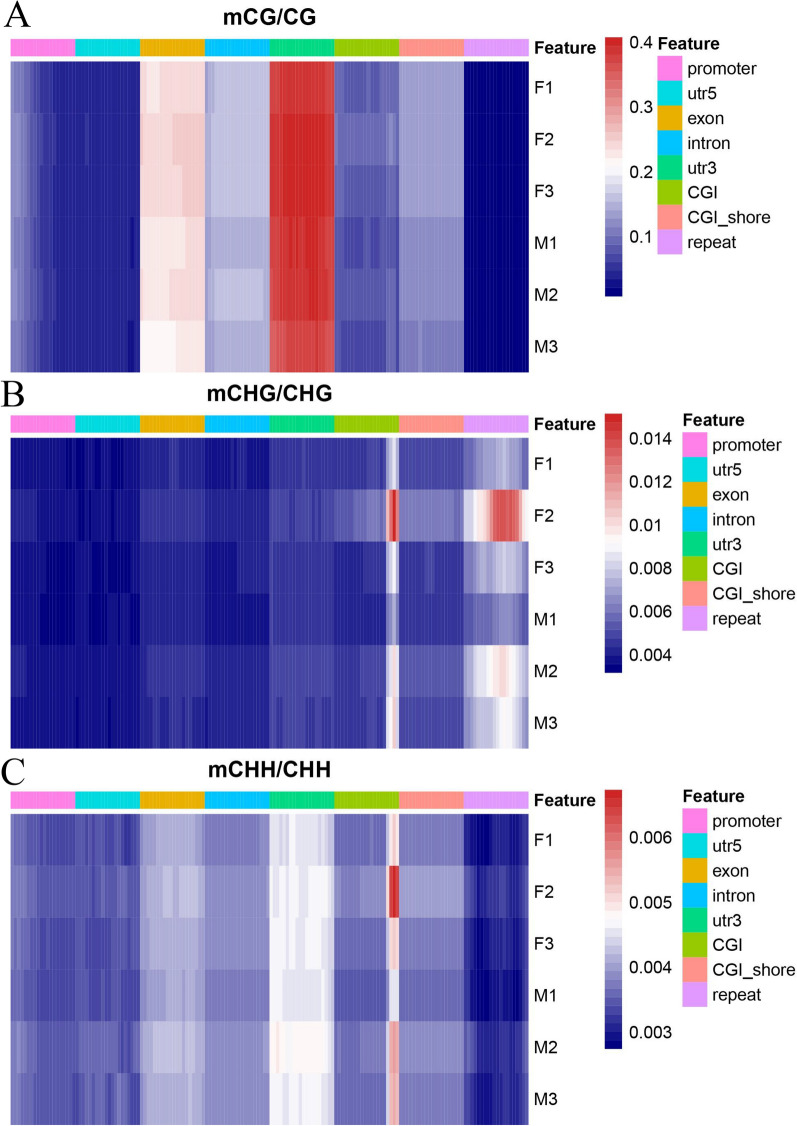
The results revealed that DNA methylation levels in H. longicornis varied by sex and sequence context (CG, CHG, and CHH). The 3' untranslated region (UTR) had the highest methylation in the CG context, followed by exons, introns, and CGI_shore regions. Female ticks generally exhibited higher methylation levels than males, particularly in gene body regions. A total of 10,460 DMRs were identified, with 5282 hypermethylated and 5178 hypomethylated. Further, GO and KEGG pathway analyses showed that differentially methylated genes (DMGs) were highly enriched in binding and metabolic pathways.
These results broaden our understanding of DNA methylation changes associated with the female and male of H. longicornis and provide an important theoretical basis for subsequent studies of epigenetic mechanisms of sex differences in ticks.
长角血蜱是一种重要的媒介,可将多种病原体传播给人类和动物。这种蜱虫的独特之处在于有两个独立的繁殖种群:两性生殖种群和孤雌生殖种群。在两性生殖种群中,雄性和雌性之间存在形态差异,雌性通常比雄性大。DNA甲基化作为一种关键的表观遗传修饰,在维持正常细胞功能、基因表达调控和胚胎发育等生物学过程中发挥着至关重要的作用。然而,长角血蜱两性生殖种群中性别分化的表观遗传机制一直被忽视。
在本研究中,利用全基因组亚硫酸氢盐测序技术探究了长角血蜱两性生殖种群中雌性和雄性蜱虫的全基因组DNA甲基化图谱。鉴定出差异甲基化区域(DMRs),随后对DMR相关基因进行基因本体(GO)和京都基因与基因组百科全书(KEGG)通路分析。
结果显示,长角血蜱的DNA甲基化水平因性别和序列上下文(CG、CHG和CHH)而异。在CG上下文中,3'非翻译区(UTR)的甲基化水平最高,其次是外显子、内含子和CGI_岸区。雌性蜱虫的甲基化水平通常高于雄性,尤其是在基因体区域。共鉴定出10460个DMRs,其中5282个为高甲基化,5178个为低甲基化。此外,GO和KEGG通路分析表明,差异甲基化基因(DMGs)在结合和代谢通路中高度富集。
这些结果拓宽了我们对长角血蜱雌雄个体相关DNA甲基化变化的理解,并为后续蜱虫性别差异表观遗传机制的研究提供了重要的理论基础。