Das Arun, Biddanda Arjun, McCoy Rajiv C, Schatz Michael C
Department of Computer Science, Johns Hopkins University, Baltimore, MD, 21218, USA.
Department of Biology, Johns Hopkins University, Baltimore, MD 21218, USA.
bioRxiv. 2025 May 14:2025.05.14.653340. doi: 10.1101/2025.05.14.653340.
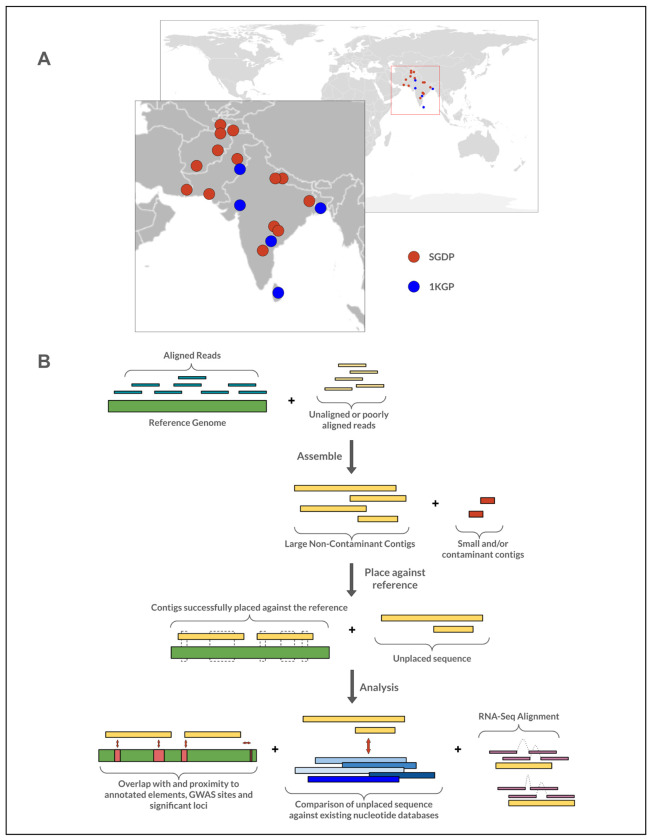
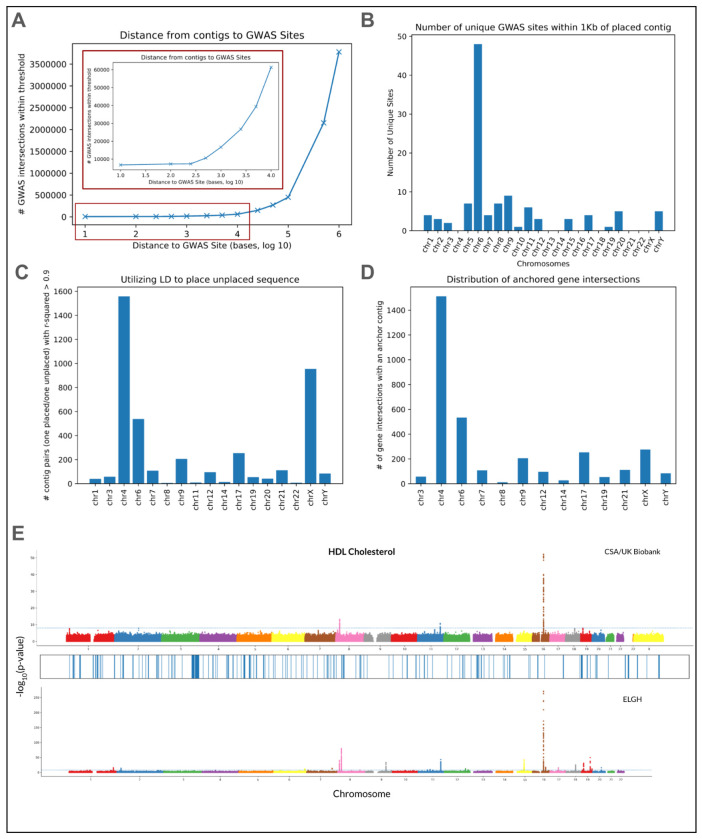
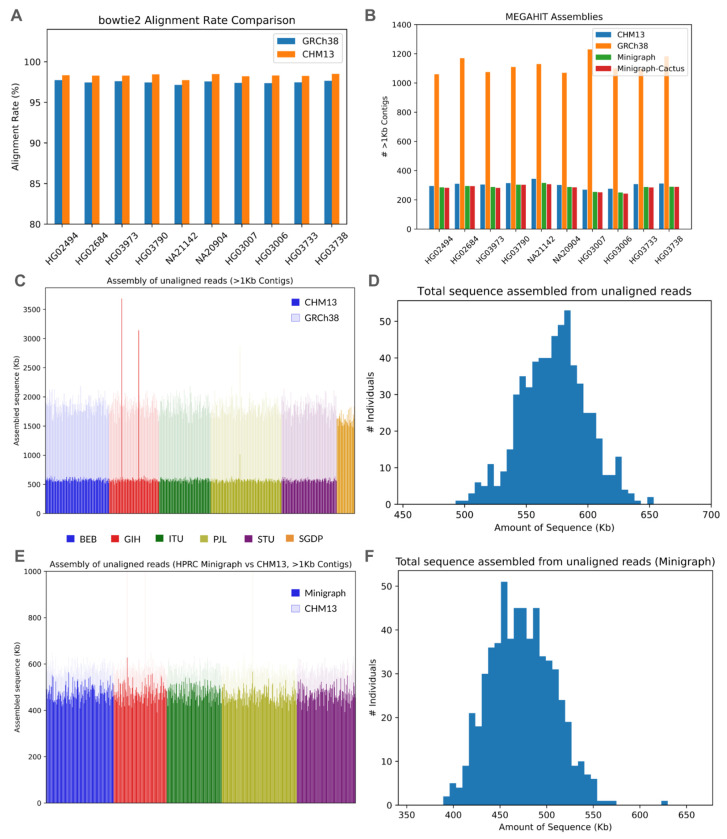
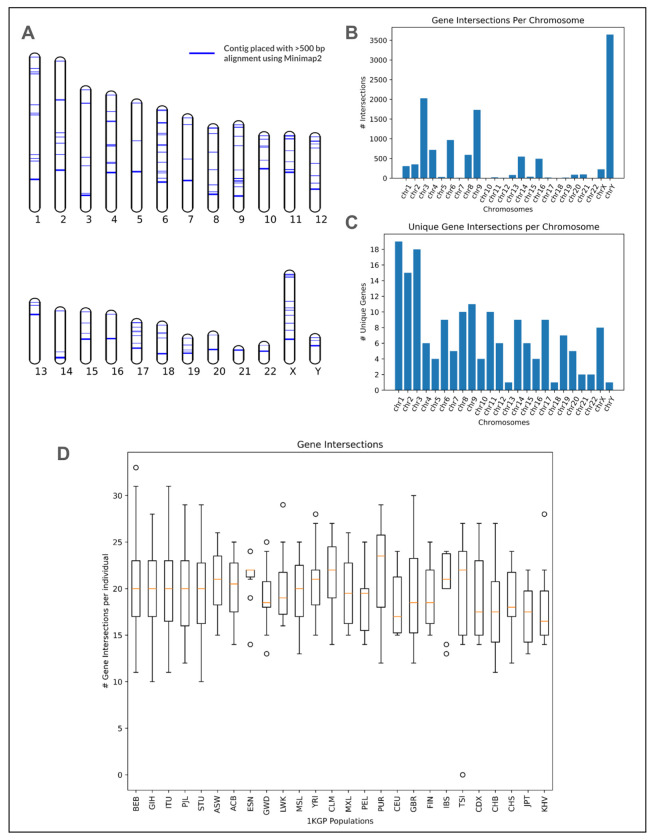
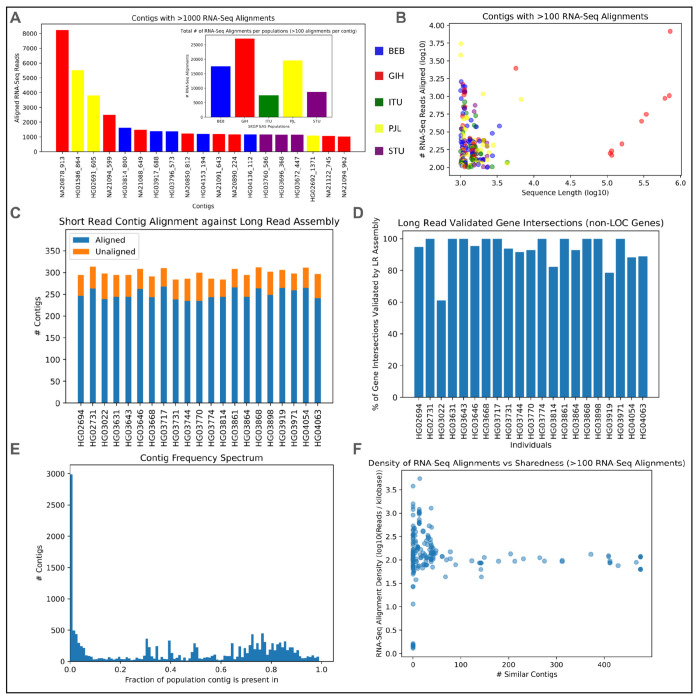
Conventional genome mapping-based approaches systematically miss genetic variation, particularly in regions that substantially differ from the reference. To explore this hidden variation, we examined unmapped and poorly mapped reads from the genomes of 640 human individuals from South Asian (SAS) populations in the 1000 Genomes Project and the Simons Genome Diversity Project. We assembled tens of megabases of non-redundant sequence in tens of thousands of large contigs, much of which is present in both SAS and non-SAS populations. We demonstrated that much of this sequence is not discovered by traditional variant discovery approaches even when using complete genomes and pangenomes. Across 20,000 placed contigs, we found 8,215 intersections with 106 protein coding genes and >15,000 placements within 1 kbp of a known GWAS hit. We used long read data from a subset of samples to validate the majority of their assembled sequences, aligned RNA-seq data to identify hundreds of unplaced contigs with transcriptional potential, and queried existing nucleotide databases to evaluate the origins of the remaining unplaced sequences. Our results highlight the limitations of even the most complete reference genomes and provide a model for understanding the distribution of hidden variation in any human population.
基于传统基因组图谱的方法会系统性地遗漏遗传变异,尤其是在与参考序列有显著差异的区域。为了探索这种隐藏的变异,我们检查了来自千人基因组计划和西蒙斯基因组多样性计划中640名南亚(SAS)人群基因组的未映射和映射不佳的读数。我们在数万个大的重叠群中组装了数十兆碱基的非冗余序列,其中许多在SAS和非SAS人群中都存在。我们证明,即使使用完整基因组和泛基因组,传统的变异发现方法也无法发现这些序列中的大部分。在20,000个已定位的重叠群中,我们发现8,215个与106个蛋白质编码基因有交集,并且在已知全基因组关联研究(GWAS)命中位点的1千碱基范围内有超过15,000个定位。我们使用来自一部分样本的长读数据来验证它们组装序列的大部分,比对RNA测序数据以识别数百个具有转录潜力的未定位重叠群,并查询现有的核苷酸数据库以评估其余未定位序列的来源。我们的结果突出了即使是最完整的参考基因组的局限性,并为理解任何人类群体中隐藏变异的分布提供了一个模型。