Michaels Jes-Rite, Iyyanar Paul P R, Husami Ammar, Vontell Andrew M, Brugmann Samantha A, Stottmann Rolf W
Steve and Cindy Rasmussen Institute for Genomic Medicine, Abigail Wexner Research Institute, Nationwide Children's Hospital, Columbus, Ohio, United States of America.
Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Cincinnati, Ohio, United States of America.
PLoS One. 2025 Jun 9;20(6):e0324803. doi: 10.1371/journal.pone.0324803. eCollection 2025.
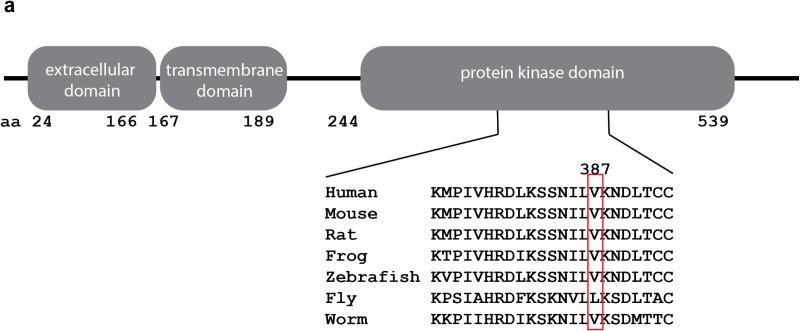
Cleft lip and cleft palate are among the most common congenital anomalies and are the result of incomplete fusion of embryonic craniofacial processes or palatal shelves, respectively. We know that genetics play a large role in these anomalies but the list of known causal genes is far from complete. As part of a larger sequencing effort of patients with congenital craniofacial anomalies, we identified a rare candidate variant in transforming growth factor beta receptor 2 (TGFBR2). This variant alters a highly conserved amino acid and is predicted to be pathogenic by a number of metrics. The family history and population genetics suggest that this specific variant would be incompletely penetrant, but this gene has been convincingly implicated in craniofacial development. In order to test the hypothesis this might be a causal variant, we used genome editing to create the orthologous variant in a new mouse model. Surprisingly, Tgfbr2V387M mice did not exhibit craniofacial anomalies or have reduced survival, suggesting Tgfbr2V387M is not a causal variant for cleft palate/ micrognathia. The discrepancy between in silico predictions and mouse phenotypes highlights the complexity of translating human genetic findings to mouse models. We expect these findings will aid in interpretation of future variants seen in TGFBR2 from ongoing sequencing of patients with congenital craniofacial anomalies.
唇腭裂是最常见的先天性畸形之一,分别是胚胎颅面突或腭突不完全融合的结果。我们知道遗传学在这些畸形中起很大作用,但已知的致病基因列表远未完整。作为对先天性颅面畸形患者进行更大规模测序工作的一部分,我们在转化生长因子β受体2(TGFBR2)中鉴定出一个罕见的候选变异。该变异改变了一个高度保守的氨基酸,并且通过多种指标预测具有致病性。家族史和群体遗传学表明该特定变异的外显率不完全,但该基因已被确凿地证明与颅面发育有关。为了检验这可能是一个致病变异的假设,我们使用基因组编辑在一个新的小鼠模型中创建了直系同源变异。令人惊讶的是,Tgfbr2V387M小鼠没有表现出颅面畸形或存活率降低,这表明Tgfbr2V387M不是腭裂/小颌畸形的致病变异。计算机预测与小鼠表型之间的差异凸显了将人类遗传发现转化为小鼠模型的复杂性。我们预计这些发现将有助于解释在先天性颅面畸形患者的持续测序中未来在TGFBR2中看到的变异。