Baylink Arden
Department of Veterinary Microbiology and Pathology, Washington State University, Pullman, Washington, USA.
bioRxiv. 2025 Jun 3:2025.06.02.657526. doi: 10.1101/2025.06.02.657526.
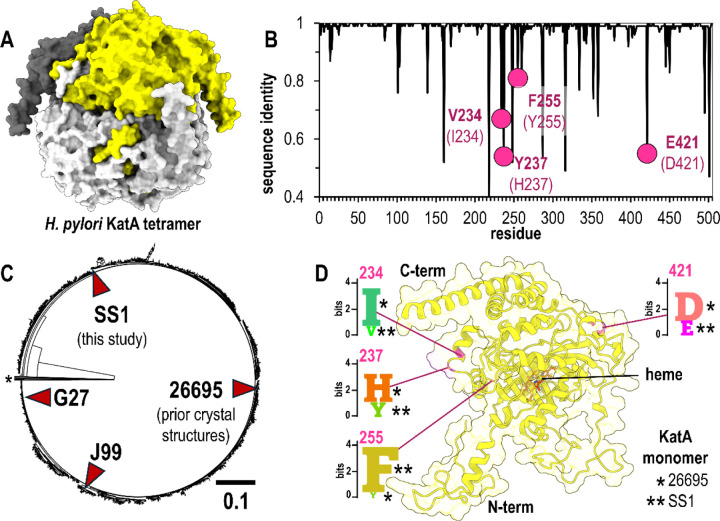
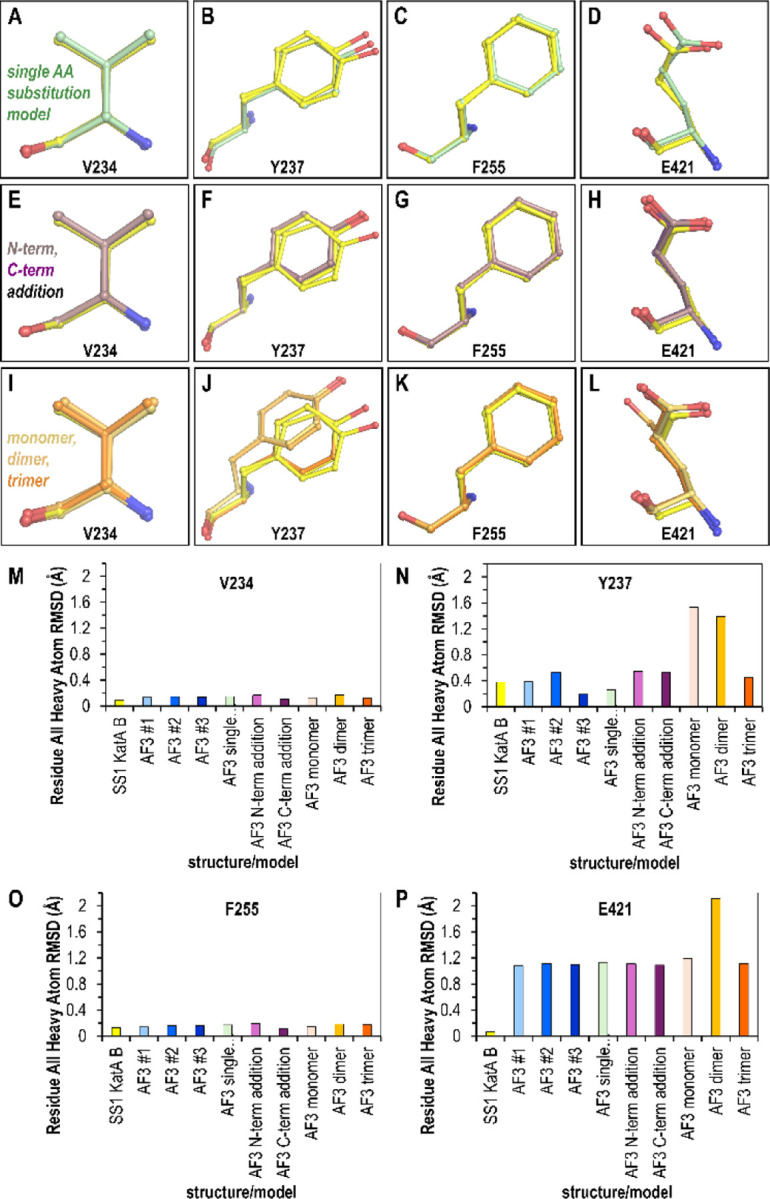
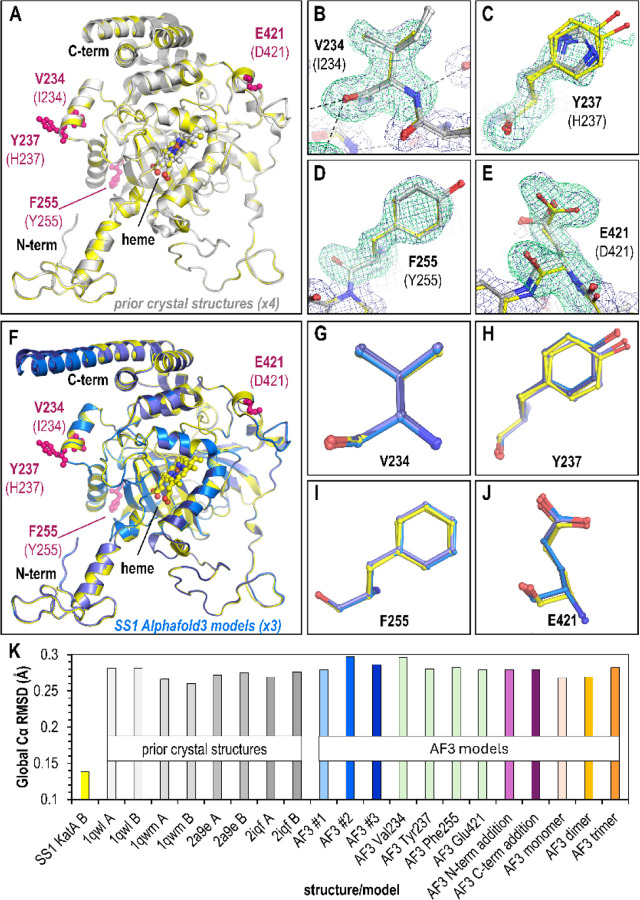
Subtle changes in protein sequence can equate to large changes in function, such as enabling pathogens to evade the immune system, hindering antibody recognition of antigens, or conferring antibiotic resistance. Even single amino acid substitutions may alter ligand binding affinity, enzymatic activity, and protein stability. Yet, due to limitations in time and resources, proteins closely related in sequence to those already characterized often remain unexamined. AlphaFold has emerged as a promising tool for protein structure prediction, though its utility in modeling single amino acid substitutions remains uncertain. In this study, we assessed AlphaFold 3's accuracy in modeling natural variants of the catalase KatA by comparing its predictions to a novel high-resolution crystal structure of KatA from strain SS1. This variant contains key substitutions at residues 234, 237, 255, and 421 relative to the well-characterized strain 26695. AlphaFold 3 models accurately reproduced the global structure and local conformations of most variant residues, with high fidelity in conservative substitutions but variable accuracy in more flexible or interface-exposed sites. We further explored how user inputs, such as incorrect oligomeric states or sequence modifications, influence prediction quality. While AlphaFold 3 consistently produced high-quality models, deviations at variant sites occurred when incorrect oligomeric states were specified. Our findings highlight both the strengths and limitations of AlphaFold 3 in modeling natural protein variants and underscore the importance of accurate user input for reliable structural predictions.
蛋白质序列的细微变化可能等同于功能上的巨大变化,例如使病原体能够逃避免疫系统、阻碍抗体对抗原的识别或赋予抗生素抗性。即使是单个氨基酸替换也可能改变配体结合亲和力、酶活性和蛋白质稳定性。然而,由于时间和资源的限制,与已表征的蛋白质序列密切相关的蛋白质往往仍未得到研究。AlphaFold已成为一种很有前景的蛋白质结构预测工具,但其在模拟单个氨基酸替换方面的效用仍不确定。在本研究中,我们通过将AlphaFold 3对过氧化氢酶KatA天然变体的预测与来自菌株SS1的KatA的新型高分辨率晶体结构进行比较,评估了其准确性。相对于特征明确的菌株26695,该变体在残基234、237、255和421处含有关键替换。AlphaFold 3模型准确地再现了大多数变体残基的整体结构和局部构象,在保守替换中具有高保真度,但在更灵活或暴露于界面的位点准确性可变。我们进一步探讨了用户输入,如错误的寡聚状态或序列修饰,如何影响预测质量。虽然AlphaFold 3始终生成高质量的模型,但当指定错误的寡聚状态时,变体位点会出现偏差。我们的研究结果突出了AlphaFold 3在模拟天然蛋白质变体方面的优势和局限性,并强调了准确的用户输入对于可靠结构预测的重要性。