Gapizov Abubakar, Syed Wajiha, Subhan Muhammad, Bibi Ruqiya, Cheema Muhammad Usairam, Mustafa Sufyan
Internal Medicine, Weill Cornell Medicine, New York Presbyterian Brooklyn Methodist Hospital, Brooklyn, USA.
Internal Medicine, King Edward Medical University, Lahore, PAK.
Cureus. 2025 May 16;17(5):e84235. doi: 10.7759/cureus.84235. eCollection 2025 May.
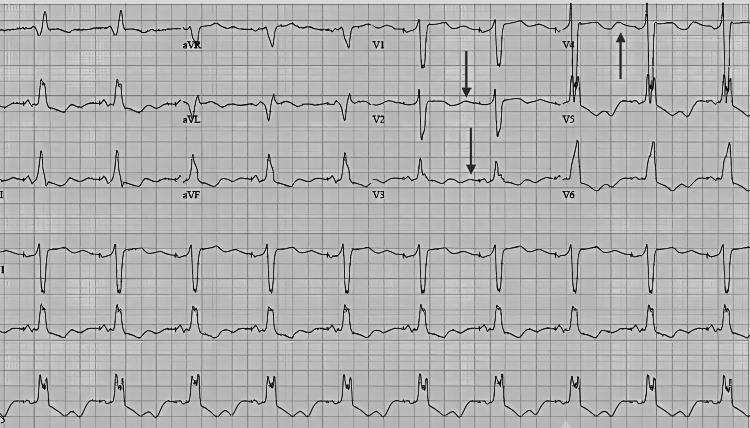
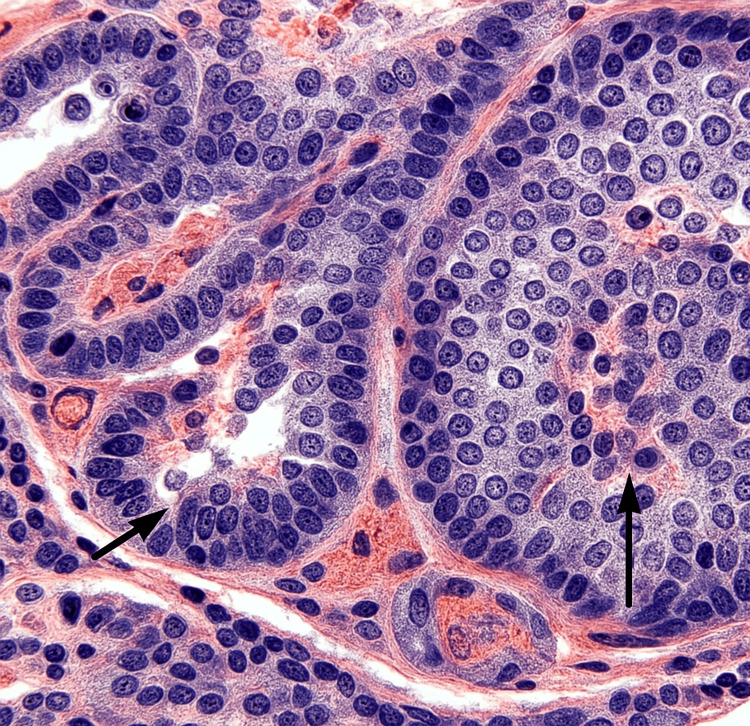
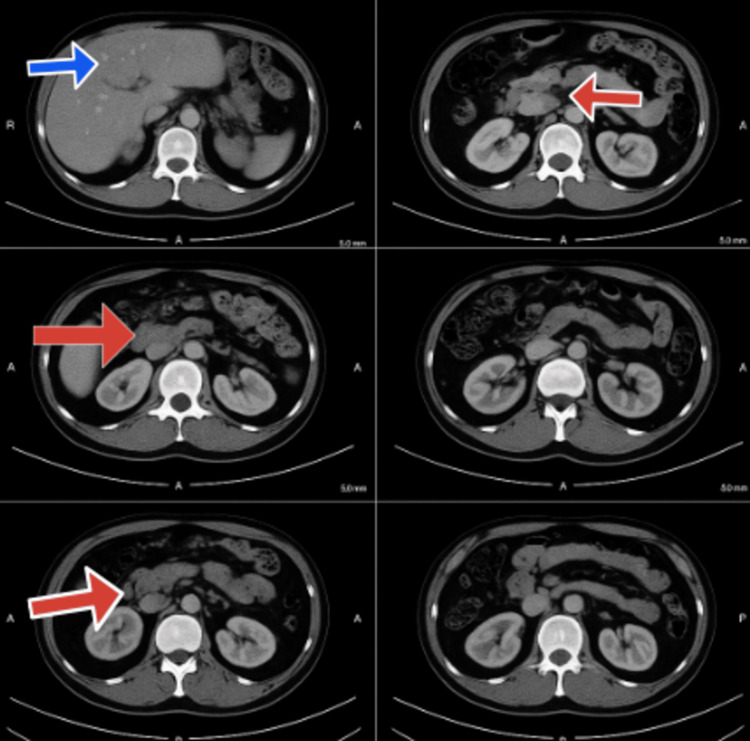
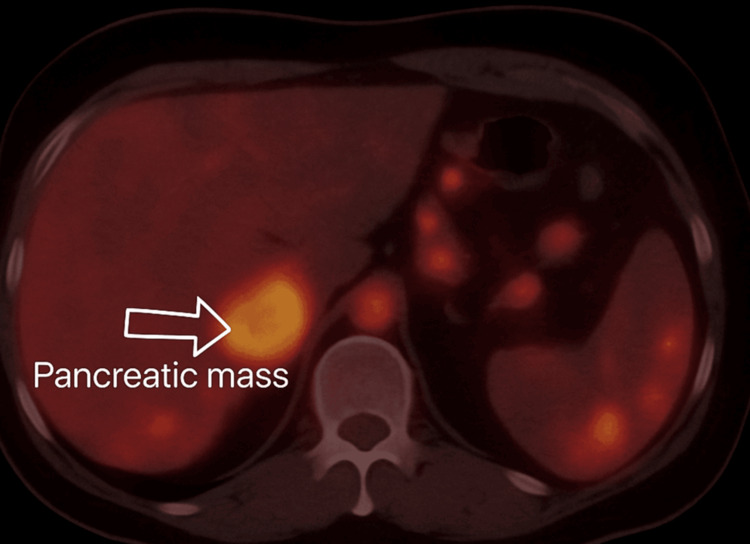
We report the first documented case of Gitelman syndrome coexisting with a metastatic pancreatic neuroendocrine tumor in a 19-year-old male, presenting with severe refractory hypokalemia (K⁺ 1.4-1.7 mmol/L), metabolic alkalosis, and hypomagnesemia. The patient's diagnostic workup revealed inappropriate renal potassium wasting (urinary K⁺ 42 mEq/L), hypocalciuria (urinary Ca²⁺/creatinine ratio <0.1), and elevated fractional chloride excretion (>1%), confirming the diagnosis of Gitelman syndrome. Imaging studies identified a somatostatin receptor-positive Grade 2 pancreatic neuroendocrine tumor (Ki-67 8%) with hepatic metastases, making surgical resection unfeasible. Management comprised high-dose potassium and magnesium supplementation, amiloride, octreotide, and everolimus. On account of disease advancement, initial treatment approaches failed, and peptide receptor radionuclide therapy remained limited for the patient owing to financial constraints. Both Gitelman syndrome and metastatic pancreatic neuroendocrine tumor posed unique challenges that required a coordinated multidisciplinary approach. This case highlights the need for malignancy to be added to the differential diagnosis of persistent electrolyte anomalies. Moreover, it emphasizes the limitations in managing double disease entities in a young individual and the insurmountable hurdles for advanced treatments like peptide receptor radionuclide therapy in underdeveloped nations. This report highlights the importance of further studying the association between the interplay of genetic syndromes (such as Gitelman syndrome) and associated neoplasms, as well as the vital coordination of complex and multidisciplinary management in rare clinical situations.
我们报告了首例有文献记载的吉特曼综合征(Gitelman syndrome)与转移性胰腺神经内分泌肿瘤并存的病例,患者为一名19岁男性,表现为严重难治性低钾血症(血钾1.4 - 1.7 mmol/L)、代谢性碱中毒和低镁血症。患者的诊断检查显示存在不适当的肾性钾丢失(尿钾42 mEq/L)、低钙尿症(尿钙/肌酐比值<0.1)以及氯化物排泄分数升高(>1%),从而确诊为吉特曼综合征。影像学检查发现了一个生长抑素受体阳性的2级胰腺神经内分泌肿瘤(Ki-67为8%)并有肝转移,使得手术切除不可行。治疗包括高剂量补充钾和镁、氨氯吡咪、奥曲肽和依维莫司。由于疾病进展,初始治疗方法失败,且由于经济限制,肽受体放射性核素治疗对该患者也有限。吉特曼综合征和转移性胰腺神经内分泌肿瘤都带来了独特的挑战,需要多学科协调方法。该病例强调在持续性电解质异常的鉴别诊断中需要考虑恶性肿瘤。此外,它凸显了在年轻个体中管理两种疾病实体的局限性以及在不发达国家中肽受体放射性核素治疗等先进治疗面临的不可逾越的障碍。本报告强调了进一步研究遗传综合征(如吉特曼综合征)与相关肿瘤之间相互作用的关联以及在罕见临床情况下复杂多学科管理的重要协调的重要性。