Hristodor Anca Manuela, Cappelli Enrico, Baldisseri Elena, Valli Roberto, Montalbano Giuseppe, Micheloni Giovanni, Porta Giovanni, Frattini Annalisa, Ravera Silvia, Fioredda Francesca, Lippi Giuseppe, Dufour Carlo, Cipolli Marco, Bezzerri Valentino
Cystic Fibrosis Center, Azienda Ospedaliera Universitaria Integrata, Verona, Italy.
Unit of Hematology, IRCCS G. Gaslini, Genoa, Italy.
Cell Death Discov. 2025 Jun 21;11(1):286. doi: 10.1038/s41420-025-02571-0.
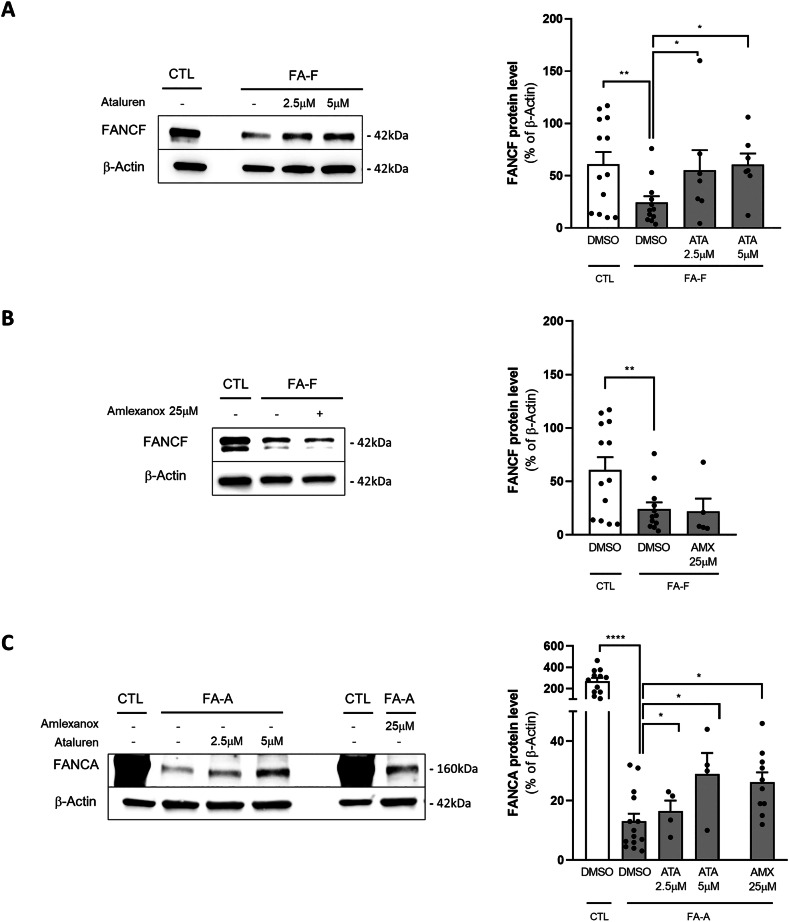
Fanconi anemia (FA) is caused by mutations affecting FANC genes involved in DNA repair, with nearly 20% of FA patients harboring nonsense mutations. Ataluren (PTC124) is a translational read-through-inducing drug (TRID) already approved in Europe that has a well-established safety profile even in pediatric patients. Amlexanox, an anti-inflammatory drug, also promotes read-through of premature stop codons caused by nonsense mutations. We compared ataluren and amlexanox in rescuing FANCA, FANCC and FANCF protein synthesis in lymphoblastoid cell lines and fibroblasts obtained from FA patients with nonsense mutations. While ataluren restored all FANC protein levels, amlexanox was partially effective only on FANCA. Notably, the rescue of FANC proteins resulted in a significant downregulation of p53. Moreover, unlike amlexanox, ataluren remarkably improved cell viability and reduced chromosomal aberrations upon exposure to genotoxic compounds. Amlexanox primarily reduced the signal transducer and activator of transcription 2 (STAT2) phosphorylation. Furthermore, FANCA-mutated fibroblasts exhibited a higher frequency of micronuclei formation as well as lower lamin B1 expression compared to their gene-edited counterpart re-expressing wild-type FANCA. Interestingly, ataluren significantly limited the generation of micronuclei in nonsense-mutated primary FANCC fibroblasts, restoring lamin B1 expression. This study represents a milestone of drug development for FA as it paves the way for clinical development of TRIDs, indicating ataluren as a promising approach to address the genetic instability and reduce the risk of malignant transformation in FA cells. Moreover, these results highlight the importance of a reliable experimental pipeline to assess whether minimal protein rescue via translational read-through can yield meaningful phenotypic rescue.
范可尼贫血(FA)是由影响参与DNA修复的FANC基因的突变引起的,近20%的FA患者携带无义突变。阿他芦醇(PTC124)是一种已在欧洲获批的通读诱导药物(TRID),即使在儿科患者中也有良好的安全性记录。抗炎症药物氨来呫诺也能促进由无义突变导致的提前终止密码子的通读。我们比较了阿他芦醇和氨来呫诺在挽救从携带无义突变的FA患者获得的淋巴母细胞系和成纤维细胞中FANCA、FANCC和FANCF蛋白合成方面的作用。虽然阿他芦醇恢复了所有FANC蛋白水平,但氨来呫诺仅对FANCA部分有效。值得注意的是,FANC蛋白的挽救导致p53显著下调。此外,与氨来呫诺不同,阿他芦醇在暴露于基因毒性化合物时显著提高了细胞活力并减少了染色体畸变。氨来呫诺主要降低了信号转导和转录激活因子2(STAT2)的磷酸化。此外,与重新表达野生型FANCA的基因编辑对应物相比,FANCA突变的成纤维细胞表现出更高频率的微核形成以及更低的核纤层蛋白B1表达。有趣的是,阿他芦醇显著限制了无义突变的原发性FANCC成纤维细胞中微核的产生,恢复了核纤层蛋白B1的表达。这项研究代表了FA药物开发的一个里程碑,因为它为TRIDs的临床开发铺平了道路,表明阿他芦醇是解决FA细胞中遗传不稳定性并降低恶性转化风险的一种有前景的方法。此外,这些结果强调了一个可靠的实验流程对于评估通过翻译通读进行的最小蛋白挽救是否能产生有意义的表型挽救的重要性。