Gillman Luciana, Condón Federico, Petroli Cesar, Rivas Mercedes
Departamento de Sistemas Agrarios y Paisajes Culturales, Centro Universitario Regional del Este, Universidad de la República, Treinta y tres y Rocha, Uruguay.
INIA Estanzuela, Unidad de Semillas y Recursos Fitogenéticos, Instituto Nacional de Investigación Agropecuaria (INIA), Colonia, Uruguay.
PLoS One. 2025 Jun 25;20(6):e0325548. doi: 10.1371/journal.pone.0325548. eCollection 2025.
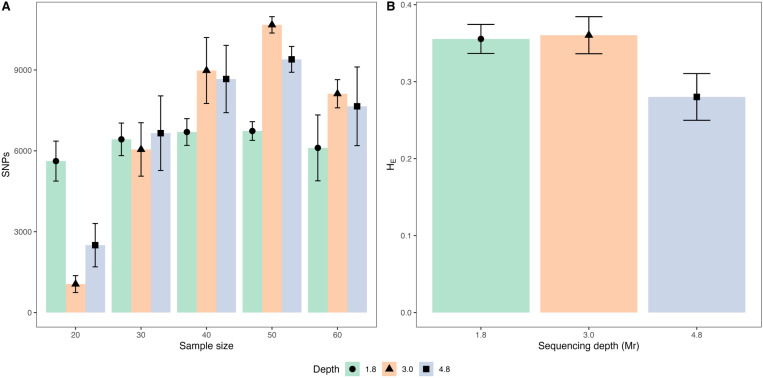
Bromus auleticus, a valuable forage grass native to the Pampa biome, is currently undergoing genetic erosion. Therefore, it is essential to assess appropriate methodologies for developing population genomic studies that will contribute to the conservation of this genetic resource. In this study, we evaluated five accessions using two genotyping strategies: individual sequencing (ind-seq) and pooled sequencing (pool-seq). To assess methodologies effectiveness, the correlation between allele frequencies calculated using each approach was investigated, as well as genetic diversity and population structure. These comparisons explicitly accounted for the potential effects of factors such as sample size, missing data, sequencing depth, and minor allele frequencies. The highest values of frequencies concordance and percentage of SNPs in common between ind-seq and pool-seq were achieved using a sample size of 30-60 plants per accession. These values were obtained with a maximum missing data threshold of 10% and a less strict minimum allele frequency threshold for pool-seq (0.01) compared to ind-seq (0.05). Pool-seq required a higher sequencing depth per accession (4.8 million reads) compared to ind-seq (0.9 million reads) to achieve similar allele frequencies. Pools of 50 individuals yielded the highest number of polymorphic sites, averaging over 9,000 per accession at a sequencing depth of 4.8 Mr. Under these conditions, pool-seq consistently resulted in an average of 0.09 higher expected heterozygosity and a 0.24 lower allelic richness compared to ind-seq in all accessions. Population structure inferred with both methodologies confirmed the outcrossing nature of B. auleticus and aligned with the geographical origin of each accession. The average inbreeding coefficient of 0.2 evidence inbreeding, which highlights the importance of conservation efforts for this valuable plant genetic resource. Based on these findings, we propose two workflows for conducting population genomics studies on Bromus auleticus.
奥氏雀麦是一种原产于潘帕斯生物群落的珍贵饲草,目前正遭受遗传侵蚀。因此,评估适当的方法以开展种群基因组研究对于保护这种遗传资源至关重要。在本研究中,我们使用两种基因分型策略对五个种质进行了评估:个体测序(ind-seq)和混合测序(pool-seq)。为了评估方法的有效性,我们研究了使用每种方法计算的等位基因频率之间的相关性,以及遗传多样性和种群结构。这些比较明确考虑了样本大小、缺失数据、测序深度和次要等位基因频率等因素的潜在影响。每个种质使用30-60株植物的样本量时,ind-seq和pool-seq之间的频率一致性和共有SNP百分比达到最高值。这些值是在最大缺失数据阈值为10%且pool-seq的最小等位基因频率阈值(0.01)比ind-seq(0.05)宽松的情况下获得的。与ind-seq(90万条 reads)相比,pool-seq每个种质需要更高的测序深度(480万条 reads)才能获得相似的等位基因频率。50个个体的混合样本产生的多态性位点数量最多,在测序深度为480万条 reads时,每个种质平均超过9000个。在这些条件下,与ind-seq相比,pool-seq在所有种质中始终导致平均预期杂合度高0.09,等位基因丰富度低0.24。两种方法推断的种群结构证实了奥氏雀麦的异交性质,并与每个种质的地理起源一致。平均近交系数为0.2表明存在近交现象,这突出了保护这种珍贵植物遗传资源的重要性。基于这些发现,我们提出了两种用于开展奥氏雀麦种群基因组研究的工作流程。