Milanovic Borko, Vijatov-Djuric Gordana, Djuretic Andrea, Kesic Jelena, Stojanovic Vesna, Jaric Milica, Ležakov Ognjen
Medical Faculty of Novi Sad, University of Novi Sad, Hajduk Veljkova 3, 21000 Novi Sad, Serbia.
Institute for Child and Youth Healthcare of Vojvodina, Hajduk Veljkova 10, 21000 Novi Sad, Serbia.
Children (Basel). 2025 May 23;12(6):672. doi: 10.3390/children12060672.
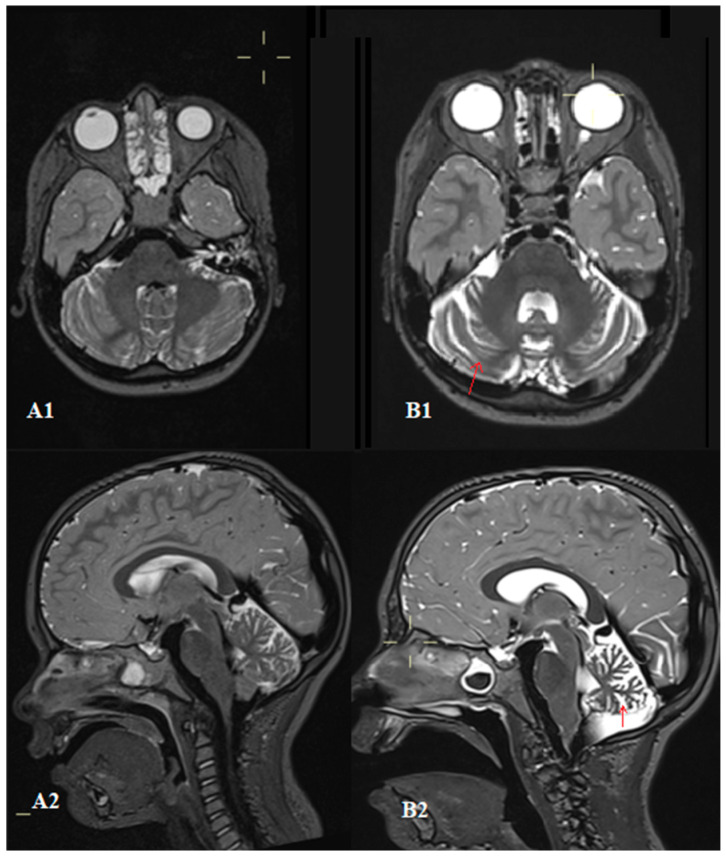
Ataxia-telangiectasia is a rare autosomal recessive disorder that is difficult to diagnose due to its unpredictable presentation. It is characterized by cerebellar degeneration, telangiectasias, immunodeficiency, frequent pulmonary infections, and tumors. Immune system abnormalities manifest as disruptions in both cellular and humoral immunity. The most common findings include decreased levels of immunoglobulin classes (IgA, IgM, IgG, and IgG subclasses) and a reduced number of T and B lymphocytes. A four-year-old girl was initially evaluated and treated for skin lesions that presented as crusts spreading across her body. She was monitored by a pulmonologist due to frequent bronchial obstructions. Over time, she developed bilateral scleral telangiectasia, saccadic eye movements, and impaired convergence. Her gait was wide-based and unstable, with truncal ataxia and a positive Romberg sign. Laboratory tests revealed decreased immunoglobulin G levels, subclass IgG4 levels, elevated alpha-fetoprotein, and a reduced number of T and B lymphocytes. Brain magnetic resonance imaging showed cerebellar atrophy. Whole-exome sequencing identified heterozygous variants c.1564-165del, p.(Glu5221lefsTer43), and c.7630-2A>C in the serine/threonine-protein kinase ATM (ataxia-telangiectasia mutated) gene, confirming the diagnosis of ataxia-telangiectasia. Following diagnosis, treatment with intravenous immunoglobulin replacement was initiated along with infection prevention and management. The goal of this case report is to raise awareness of the atypical initial presentation that may lead to a diagnostic delay. We emphasize the importance of considering ataxia-telangiectasia in the differential diagnosis, even when classical neurological signs are not yet evident.
共济失调毛细血管扩张症是一种罕见的常染色体隐性疾病,因其临床表现不可预测而难以诊断。其特征为小脑变性、毛细血管扩张、免疫缺陷、频繁肺部感染和肿瘤。免疫系统异常表现为细胞免疫和体液免疫均受到破坏。最常见的发现包括免疫球蛋白类别(IgA、IgM、IgG和IgG亚类)水平降低以及T和B淋巴细胞数量减少。一名4岁女童最初因全身出现结痂的皮肤病变接受评估和治疗。由于频繁出现支气管阻塞,她由一名肺科医生进行监测。随着时间推移,她出现了双侧巩膜毛细血管扩张、眼球跳动性眼球运动和集合功能受损。她的步态呈宽基底且不稳定,伴有躯干共济失调和Romberg征阳性。实验室检查显示免疫球蛋白G水平降低、IgG4亚类水平升高、甲胎蛋白升高以及T和B淋巴细胞数量减少。脑部磁共振成像显示小脑萎缩。全外显子测序在丝氨酸/苏氨酸蛋白激酶ATM(共济失调毛细血管扩张症突变)基因中鉴定出杂合变异c.1564 - 165del、p.(Glu5221lefsTer43)和c.7630 - 2A>C,从而确诊为共济失调毛细血管扩张症。确诊后,开始进行静脉注射免疫球蛋白替代治疗,并同时进行感染预防和管理。本病例报告的目的是提高对可能导致诊断延迟的非典型初始表现的认识。我们强调在鉴别诊断中考虑共济失调毛细血管扩张症的重要性,即使经典的神经学体征尚不明显。