Zhang Lei, Hu Lan, Tan Li, Zhang Zhenjie, Chen Mengying, Gan Wenbo, Chen Li, Zou Yan, Wang Shi, Pang Yu, Fan Zhenxin, Liu Junjie
Guang'an People's Hospital, Guan'an, China.
Department of Emergency Medicine, West China Hospital, West China School of Nursing, Sichuan University, Chengdu, China.
Front Med (Lausanne). 2025 Jul 16;12:1618947. doi: 10.3389/fmed.2025.1618947. eCollection 2025.
Immunoglobulin A nephropathy (IgAN) and membranous nephropathy (MN) are among the most common forms of primary glomerular diseases, with a rising global incidence. Despite their clinical importance, the underlying pathogenesis of these diseases and the development of reliable non-invasive diagnostic tools remain inadequately understood. Accumulating evidence suggests that gut microbiota and its associated metabolites may play a crucial role in the development of kidney diseases via the gut-kidney axis. However, comprehensive studies integrating both microbiome and metabolomic data in IgAN and MN are still limited.
In this study, we performed integrated metagenomic sequencing and untargeted metabolomic profiling to investigate alterations in gut microbial composition and systemic metabolic changes associated with IgAN and MN. Fecal samples were collected from 24 patients with IgAN, 20 patients with MN, and 17 healthy controls. Microbial diversity and composition were assessed using metagenomic analysis, while metabolic profiles were evaluated through untargeted LC -MS-based metabolomics. Multivariate statistical analyses and biomarker modeling were employed to identify discriminative features and evaluate diagnostic performance. Microbiota-metabolite correlation networks were constructed to explore potential mechanistic links.
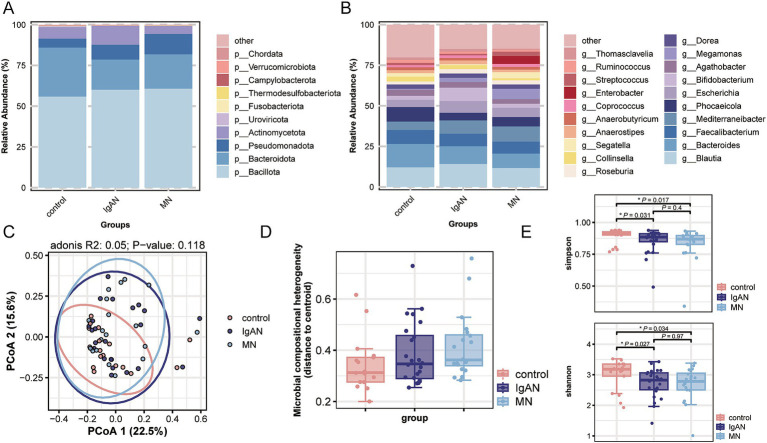
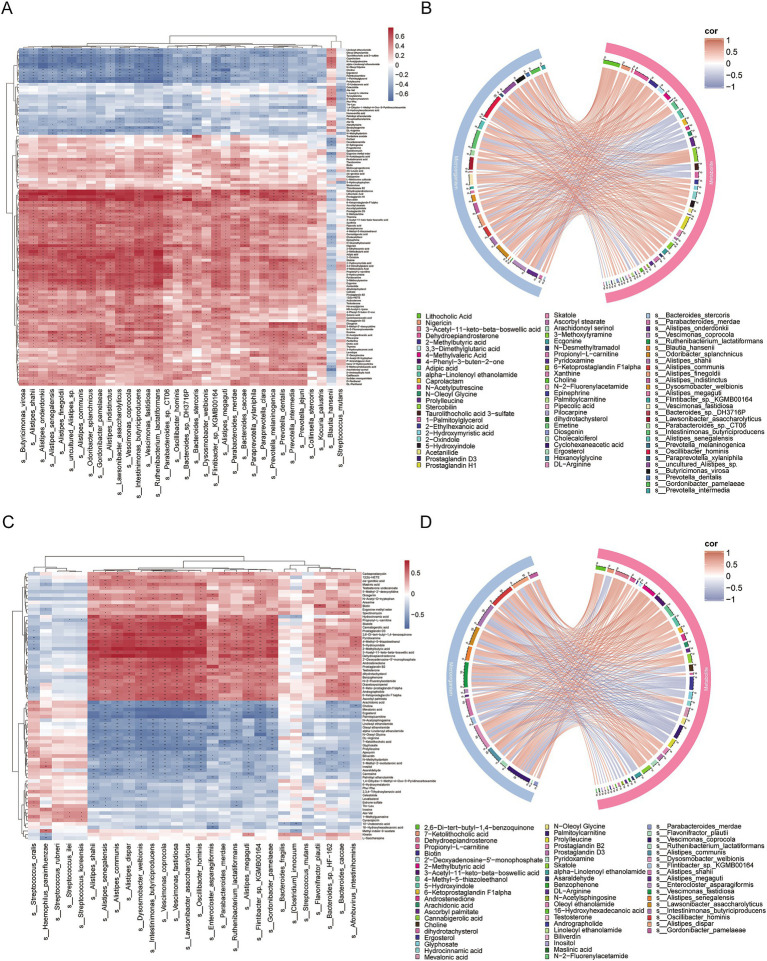
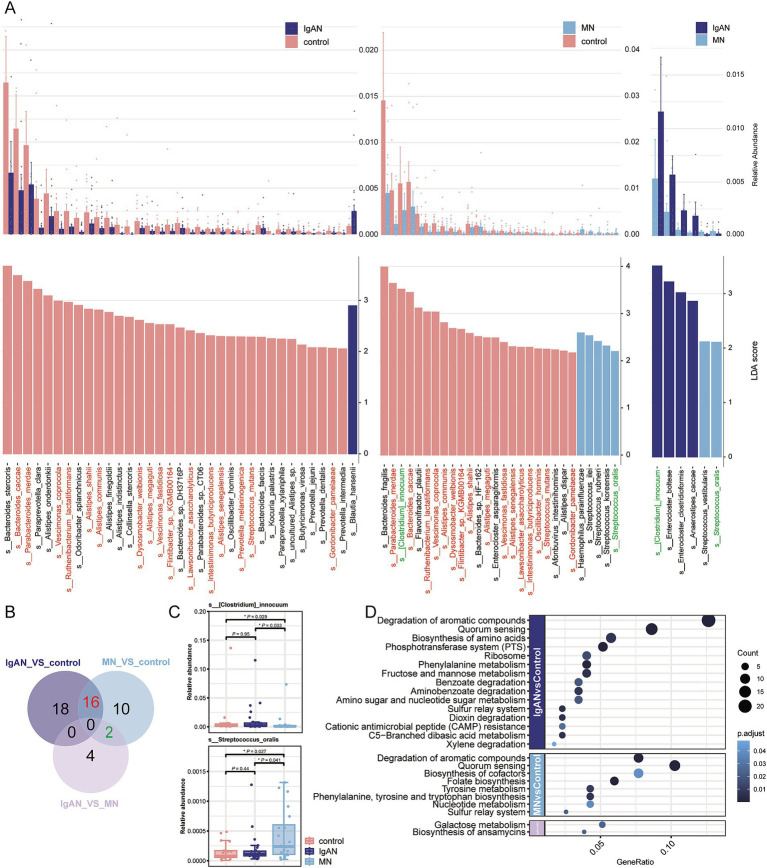
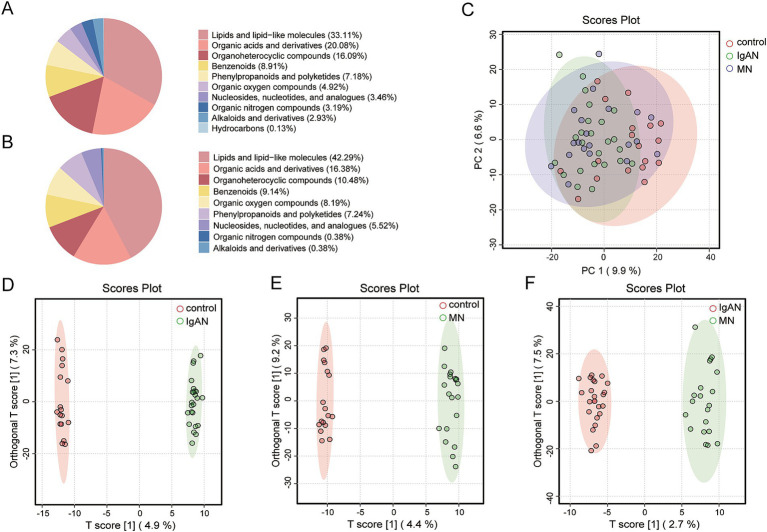
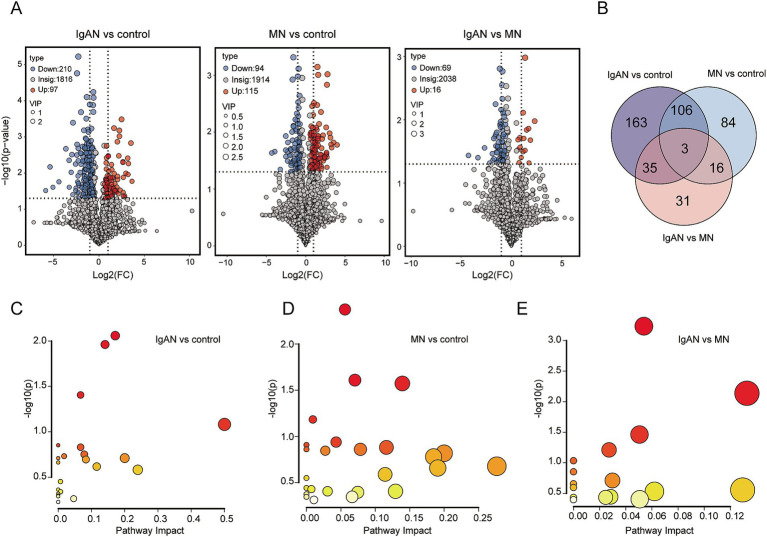
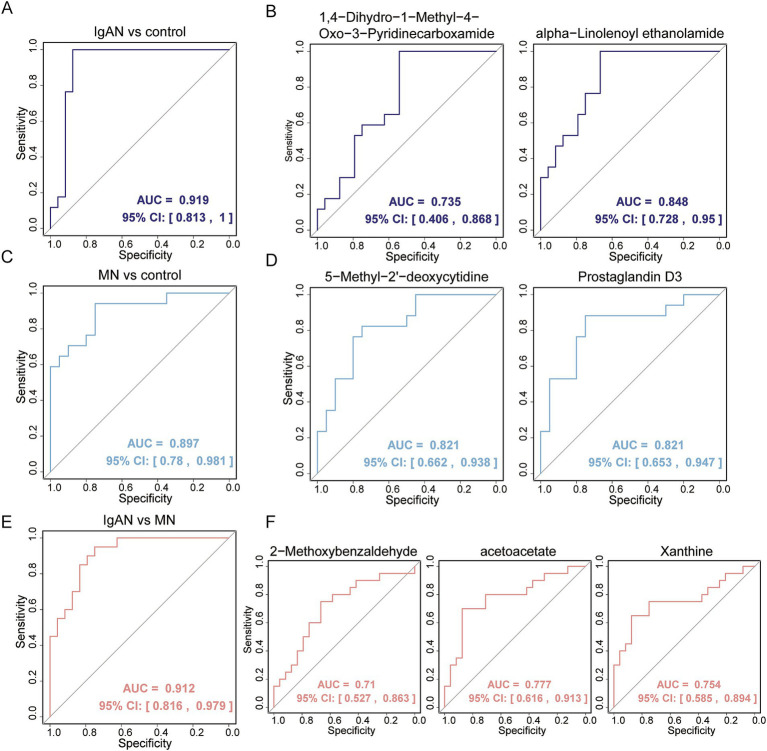
Metagenomic analysis showed that both the IgAN and MN groups had significantly reduced α-diversity. Although β-diversity analysis did not reveal significant differences between the three groups, the IgAN and MN groups exhibited higher sample dispersion than the control group. Notably, both IgAN and MN patients showed a decrease in the abundance of certain specific microbial taxa. A total of 34 and 28 differentially abundant microbial species were identified in IgAN and MN, respectively, compared to healthy controls, with 16 taxa consistently downregulated in both disease groups. Notably, was significantly enriched in the MN group, while was markedly depleted. Metabolomic profiling identified 307 and 209 differentially abundant metabolites in IgAN and MN, respectively. Dipeptides (e.g., prolylleucine) were consistently upregulated, while the levels of certain short-chain fatty acids (SCFA) were reduced. Multivariate biomarker models demonstrated excellent diagnostic performance, achieving area under the curve (AUC) of 0.919 (IgAN vs. control), 0.897 (MN vs. control) and 0.912 (IgAN vs. MN), surpassing individual metabolite markers.
Our findings highlight significant alterations in gut microbial composition and systemic metabolite profiles in both IgAN and MN patients compared to healthy individuals. The consistent reduction in microbial diversity and SCFA-producing taxa, along with characteristic changes in metabolic signatures, supports the involvement of the gut-kidney axis in disease pathogenesis. The diagnostic models developed in this study provide promising non-invasive biomarkers for distinguishing IgAN and MN with high accuracy. These results contribute novel insights into the microbe-metabolite interplay in glomerular diseases and offer potential targets for future diagnostic and therapeutic strategies.
免疫球蛋白A肾病(IgAN)和膜性肾病(MN)是原发性肾小球疾病最常见的形式,在全球发病率呈上升趋势。尽管它们具有临床重要性,但这些疾病的潜在发病机制以及可靠的非侵入性诊断工具的开发仍未得到充分了解。越来越多的证据表明,肠道微生物群及其相关代谢产物可能通过肠-肾轴在肾脏疾病的发展中起关键作用。然而,整合IgAN和MN中微生物组和代谢组数据的综合研究仍然有限。
在本研究中,我们进行了宏基因组测序和非靶向代谢组分析,以研究与IgAN和MN相关的肠道微生物组成变化和全身代谢变化。收集了24例IgAN患者、20例MN患者和17名健康对照的粪便样本。使用宏基因组分析评估微生物多样性和组成,同时通过基于非靶向液相色谱-质谱的代谢组学评估代谢谱。采用多变量统计分析和生物标志物建模来识别判别特征并评估诊断性能。构建微生物群-代谢物相关网络以探索潜在的机制联系。
宏基因组分析表明,IgAN组和MN组的α多样性均显著降低。尽管β多样性分析未显示三组之间存在显著差异,但IgAN组和MN组的样本离散度高于对照组。值得注意的是,IgAN和MN患者的某些特定微生物类群丰度均下降。与健康对照相比,IgAN和MN分别鉴定出34种和28种差异丰富的微生物物种,其中16个分类群在两个疾病组中均持续下调。值得注意的是,[具体物种]在MN组中显著富集,而[具体物种]明显减少。代谢组分析分别在IgAN和MN中鉴定出307种和209种差异丰富的代谢物。二肽(如脯氨酰亮氨酸)持续上调,而某些短链脂肪酸(SCFA)水平降低。多变量生物标志物模型显示出优异的诊断性能,IgAN与对照组比较的曲线下面积(AUC)为0.919,MN与对照组比较为0.897,IgAN与MN比较为0.912,超过了单个代谢物标志物。
我们的研究结果突出了与健康个体相比,IgAN和MN患者肠道微生物组成和全身代谢谱的显著变化。微生物多样性和产生SCFA的分类群的持续减少,以及代谢特征的特征性变化,支持肠-肾轴参与疾病发病机制。本研究中开发的诊断模型为高精度区分IgAN和MN提供了有前景的非侵入性生物标志物。这些结果为肾小球疾病中微生物-代谢物相互作用提供了新的见解,并为未来的诊断和治疗策略提供了潜在靶点。