Coldbeck-Shackley Rosa C, Lawrence Andrew, McMillan Mark, Selway Caitlin A, Papanicolas Lito, Turra Mark, Marshall Helen, Leong Lex E X
Microbiology and Infectious Diseases, SA Pathology, Adelaide 5000, Australia.
Vaccinology and Immunology Research Trials Unit, Women's and Children's Health Network, Adelaide 5000, Australia.
Microb Genom. 2025 Aug;11(8). doi: 10.1099/mgen.0.001464.
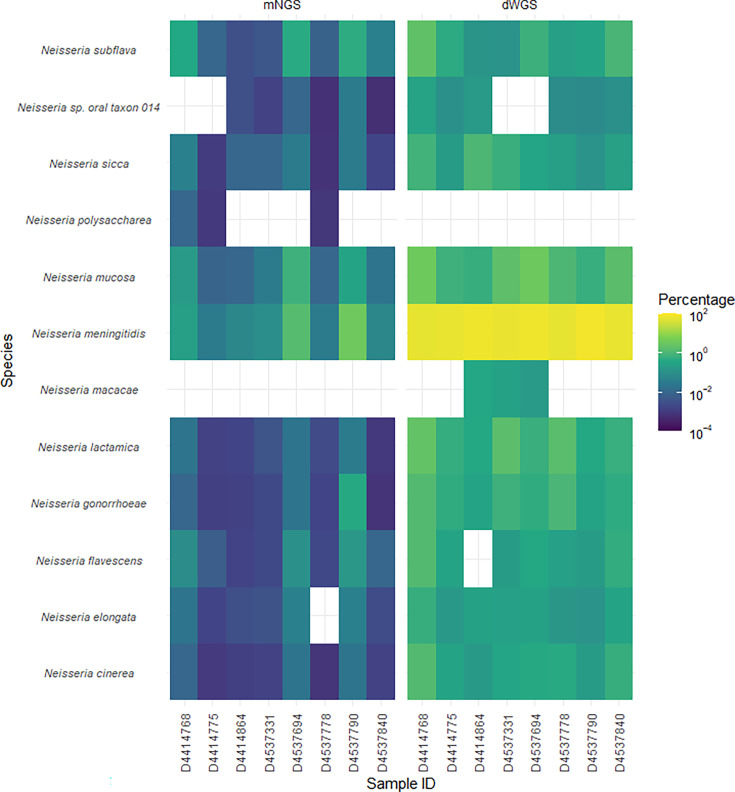
Oropharyngeal carriage of (.) is a prerequisite for invasive meningococcal disease. As such, genomic surveillance of disease-causing carriage strains can inform targeted public health responses. However, whole-genome sequencing (WGS) from isolates is often precluded due to the high rates of culture failure for . samples collected in carriage studies. This study outlines an alternative method to sequence . directly from oropharyngeal specimens that enables high-resolution molecular fine typing.We performed direct probe-capture enrichment WGS (dWGS) of . on oropharyngeal specimens from the 'B part of it' South Australian and 'B part of it NT' Northern Territory meningococcal carriage studies (NCT03089086 and NCT04398849). Sequences were analysed using currently available bioinformatic tools, including the characterization of genogroup, multi-locus sequence typing (MLST), Antigen Sequence Typing (BAST), and type.Sensitivity of dWGS typing compared to WGS for genogroup, MLST, , and BAST schemes was 88.89%, 72.22%, 100%, 94.44% and 88.24%, respectively. Genogroup and type were more reliably characterized in unculturable samples compared to the other typing schemes assessed. Factors that influenced accurate fine typing included the amount and proportion of . sequences, and the proportion of other species in enriched sequencing libraries. An alternative phylogenetic method (phylotyping) correctly predicted the clonal complex for 93.46% of the samples assessed. These results demonstrate that dWGS enables high-resolution molecular fine typing and can be applied to unculturable samples in . carriage studies.
[脑膜炎奈瑟菌]的口咽携带是侵袭性脑膜炎球菌病的先决条件。因此,对致病携带菌株进行基因组监测可为有针对性的公共卫生应对措施提供信息。然而,由于在携带研究中采集的[脑膜炎奈瑟菌]样本培养失败率很高,通常无法从分离株进行全基因组测序(WGS)。本研究概述了一种直接从口咽标本对[脑膜炎奈瑟菌]进行测序的替代方法,该方法能够进行高分辨率分子精细分型。我们对南澳大利亚州“[研究名称]的B部分”和北领地“[研究名称]的B部分”脑膜炎球菌携带研究(NCT03089086和NCT04398849)的口咽标本进行了[脑膜炎奈瑟菌]的直接探针捕获富集WGS(dWGS)。使用当前可用的生物信息学工具对序列进行分析,包括基因群的表征、多位点序列分型(MLST)、[血清群抗原序列分型(BAST)]、[血清型]和[亚型]分型。与WGS相比,dWGS在基因群、MLST、[血清群抗原序列分型(BAST)]、[血清型]和[亚型]分型方案中的分型敏感性分别为88.89%、72.22%、100%、94.44%和88.24%。与评估的其他分型方案相比,在不可培养的样本中,基因群和[血清型]分型的特征更可靠。影响准确精细分型的因素包括[脑膜炎奈瑟菌]序列的数量和比例,以及富集测序文库中其他[奈瑟菌]物种的比例。一种替代的系统发育方法(系统发育分型)正确预测了93.46%评估样本的克隆复合体。这些结果表明,dWGS能够进行高分辨率分子精细分型,可应用于[脑膜炎奈瑟菌]携带研究中的不可培养样本。