Cen Ziyan, Ge Pingping, Chen Yuhe, Zhang Ting, Wang Peiyao, Hu Lingwei, Wu Benqing, Huang Xinwen
Department of Genetics and Metabolism, Children's Hospital of Zhejiang University School of Medicine, National Clinical Research Center for Child Health, No. 3333 Binsheng Road, Binjiang District, Hangzhou, 310053, Zhejiang, China.
Children's Medical Center, University of the Chinese Academy of Sciences-Shenzhen Hospital, Shenzhen, 518106, Guangdong, China.
Orphanet J Rare Dis. 2025 Aug 13;20(1):432. doi: 10.1186/s13023-025-03985-w.
Urea cycle disorders (UCDs) are a group of rare genetic metabolic disorders characterized by hyperammonemia, which can lead to neurological damage, systemic complications, and even death. Understanding UCDs' clinical features and progression in the Chinese population will fill research gaps and benefit patients globally.
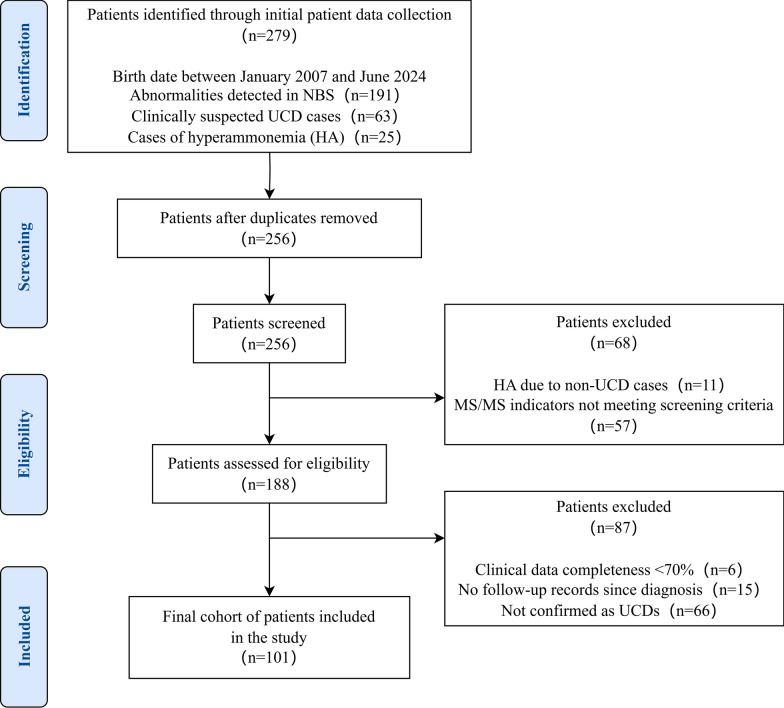
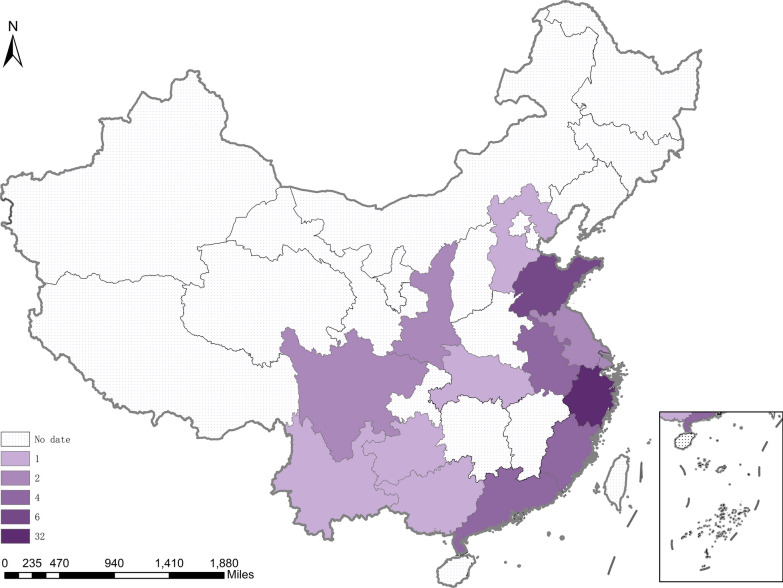
This retrospective study evaluated the clinical, biochemical, genetic characteristics, and long-term outcomes in 101 Chinese patients with six subtypes of UCDs between 2007 and 2024. Data were collected from medical records and analyzed.
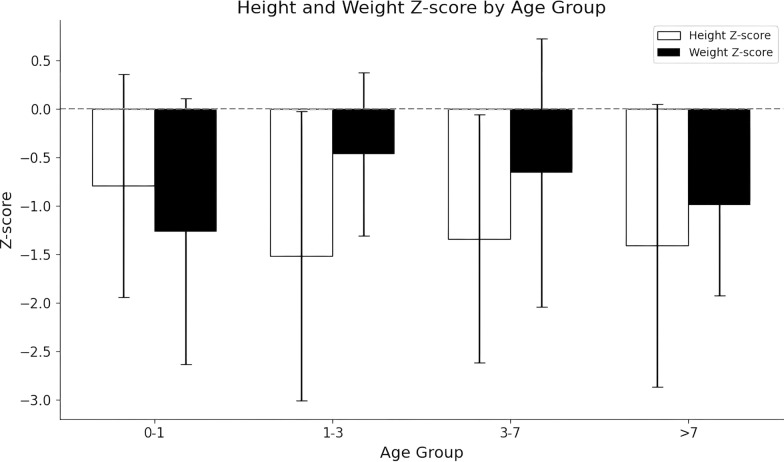
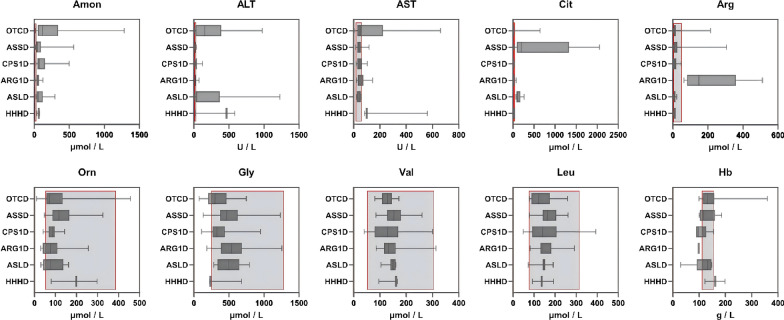
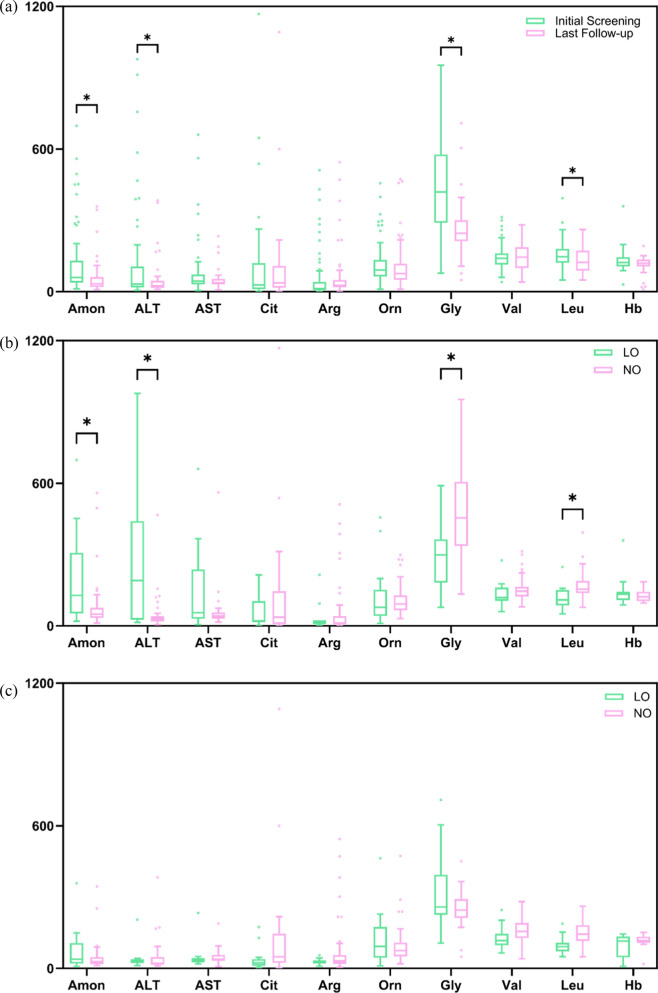
The overall survival rate was 93.0% among UCD patients. An equal gender ratio was observed in ornithine transcarbamylase deficiency. Newborn screening (NBS) was conducted in this cohort, and 57.0% of patients were diagnosed through NBS. Neurological and gastrointestinal symptoms were the most common. Symptoms often appeared within the first year, especially in the first month. Arginine was the most frequently used treatment, with glycerol phenylbutyrate often used as a nitrogen scavenger in severe cases. Biochemical analysis showed subtype-specific differences, including notable declines in leucine and glycine on low-protein diets. Genetic analysis revealed a wide distribution of mutations, with few hotspots and 17 newly identified mutations. Clinically diagnosed patients had worse outcomes than those diagnosed via newborn screening.
This study is the first to describe the clinical features and long-term outcomes of UCDs in a large sample of Chinese patients, highlighting the importance of newborn screening for early diagnosis and improved treatment outcomes.
尿素循环障碍(UCDs)是一组罕见的遗传性代谢障碍,其特征为高氨血症,可导致神经损伤、全身并发症甚至死亡。了解UCDs在中国人群中的临床特征和病情发展将填补研究空白,并使全球患者受益。
这项回顾性研究评估了2007年至2024年间101例患有六种UCDs亚型的中国患者的临床、生化、遗传特征及长期预后。数据从病历中收集并进行分析。
UCD患者的总体生存率为93.0%。在鸟氨酸转氨甲酰酶缺乏症中观察到性别比例相等。该队列进行了新生儿筛查(NBS),57.0%的患者通过NBS确诊。神经和胃肠道症状最为常见。症状通常在第一年出现,尤其是在第一个月。精氨酸是最常用的治疗药物,在严重病例中甘油苯丁酸常作为氮清除剂使用。生化分析显示各亚型存在特异性差异,包括低蛋白饮食时亮氨酸和甘氨酸显著下降。遗传分析揭示了突变的广泛分布,热点较少,新发现了17种突变。临床诊断的患者预后比通过新生儿筛查诊断的患者更差。
本研究首次描述了大量中国患者中UCDs的临床特征和长期预后,强调了新生儿筛查对早期诊断和改善治疗效果的重要性。