Liu Chunjiang, Wang Yuan, Zhou Lina, Cai Feifei, Tang Xiaoqi, Wang Liying, Zhang Xiang
Department of General Surgery, Division of Vascular Surgery, Shaoxing People's Hospital (The First Affiliated Hospital, Shaoxing University), Shaoxing, China.
Department of Intervention Vascular, Hefei Hospital Affiliated to Anhui Medical University, Hefei, China.
Front Genet. 2025 Jul 30;16:1546315. doi: 10.3389/fgene.2025.1546315. eCollection 2025.
Sjögren's syndrome (SS) is an autoimmune disorder impacting exocrine glands, while peripheral atherosclerosis (PA) demonstrates a close link to inflammation. Despite a notable rise in atherosclerosis risk among SS patients in prior investigations, the precise mechanisms remain elusive.
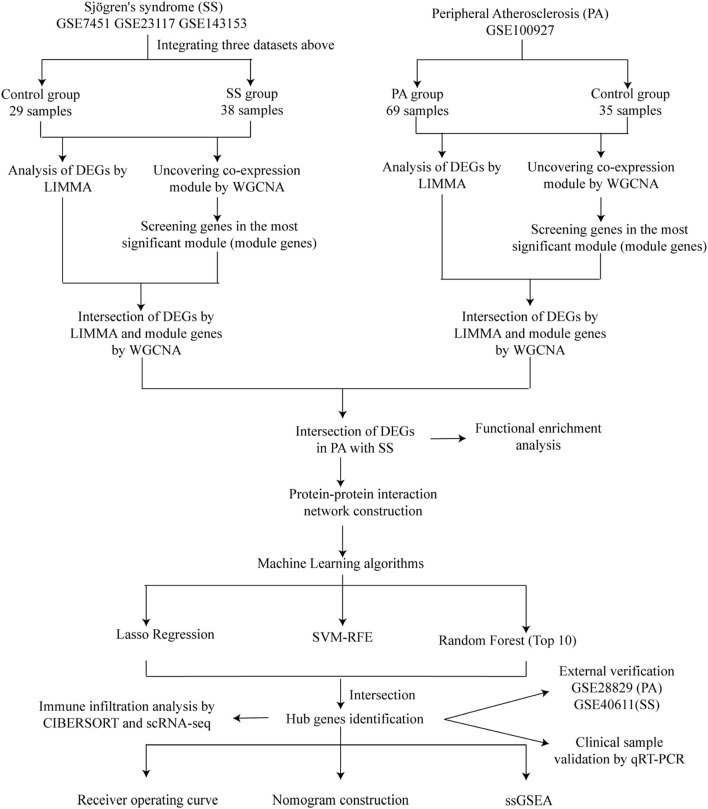
A comprehensive analysis was conducted on seven microarray datasets (GSE7451, GSE23117, GSE143153, GSE28829, GSE100927, GSE159677, and GSE40611). The LIMMA package, in conjunction with weighted gene co-expression network analysis (WGCNA), provides a robust method for identifying differentially expressed genes (DEGs) associated with peripheral atherosclerosis (PA) in Sjögren's syndrome (SS). Subsequently, machine learning algorithms and protein-protein interaction (PPI) network analysis were employed to further investigate potential predictive genes. These findings were utilized to construct a nomogram and a receiver operating characteristic (ROC) curve, which assessed the predictive accuracy of these genes in PA patients with SS. Additionally, extensive analyses of immune cell infiltration and single-sample gene set enrichment analysis (ssGSEA) were conducted to elucidate the underlying biological mechanisms.
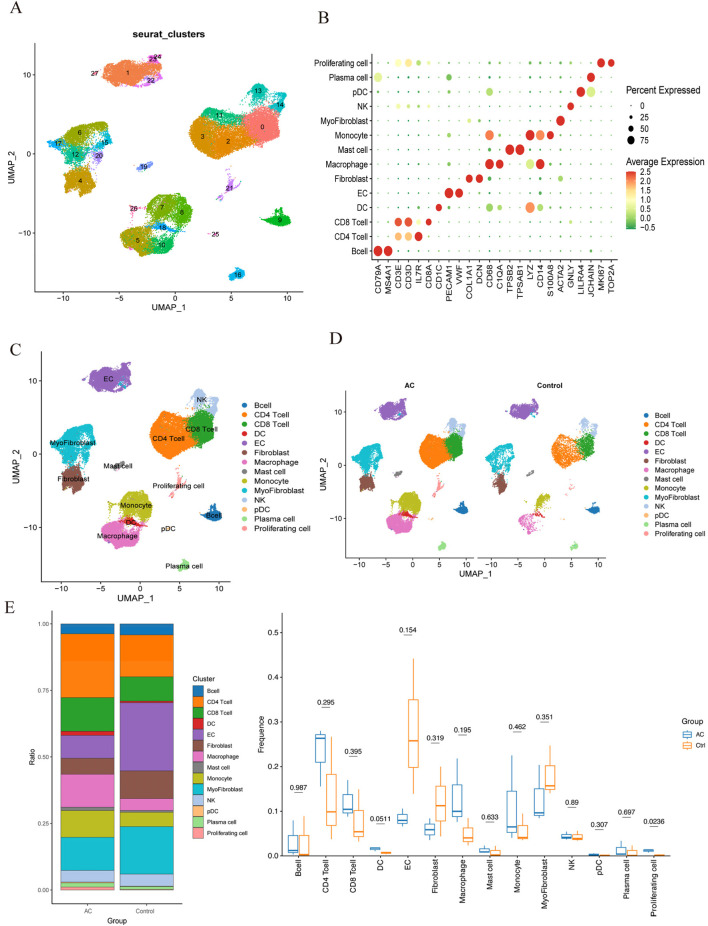
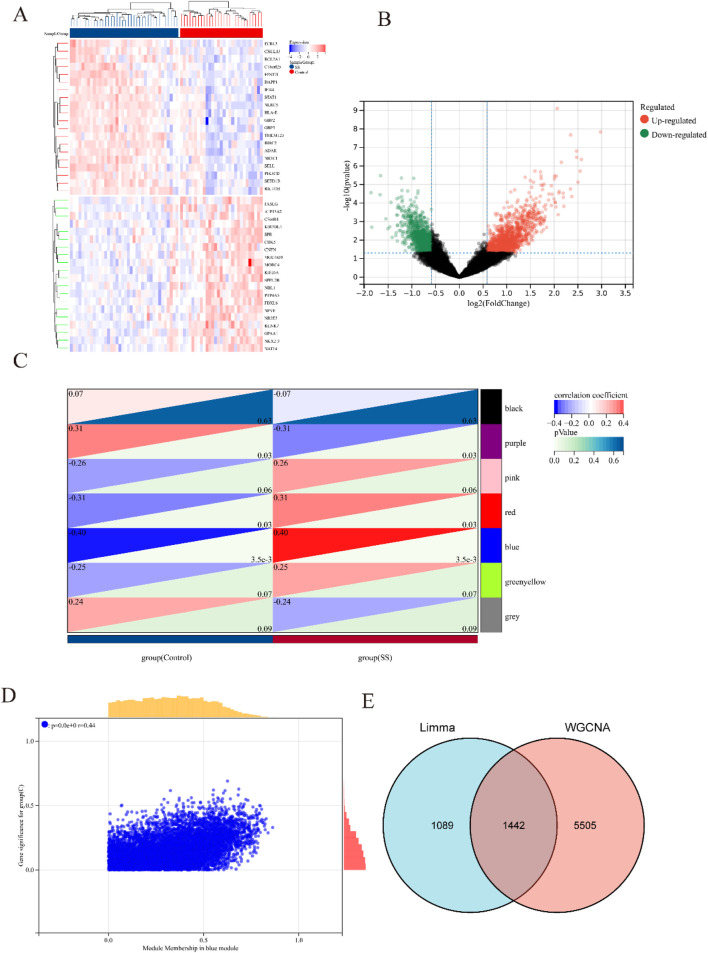
Using the LIMMA package and WGCNA, 135 DEGs associated with PA in SS were identified. PPI network analysis revealed 17 candidate hub genes. The intersection of gene sets identified by three distinct machine learning algorithms highlighted CCL4, CSF1R, and MX1 as key DEGs. ROC analysis and nomogram construction demonstrated their high predictive accuracy (AUC: 0.971, 95% CI: 0.941-1.000). Analysis of immune cell infiltration showed a significant positive correlation between these hub genes and dysregulated immune cells. Additionally, ssGSEA provided critical biological insights into the progression of PA in SS.
This study systematically identified three promising hub genes (CCL4, CSF1R, and MX1) and developed a nomogram for predicting PA in SS. Analysis of immune cell infiltration demonstrated that dysregulated immune cells significantly contribute to the progression of PA. Additionally, ssGSEA analysis offered important insights into the mechanisms by which SS leads to PA.
干燥综合征(SS)是一种影响外分泌腺的自身免疫性疾病,而外周动脉粥样硬化(PA)与炎症密切相关。尽管先前的研究表明SS患者的动脉粥样硬化风险显著增加,但其确切机制仍不清楚。
对七个微阵列数据集(GSE7451、GSE23117、GSE143153、GSE28829、GSE100927、GSE159677和GSE40611)进行了综合分析。LIMMA软件包与加权基因共表达网络分析(WGCNA)相结合,为识别干燥综合征(SS)中外周动脉粥样硬化(PA)相关的差异表达基因(DEG)提供了一种强大的方法。随后,采用机器学习算法和蛋白质-蛋白质相互作用(PPI)网络分析来进一步研究潜在的预测基因。这些结果被用于构建列线图和受试者工作特征(ROC)曲线,以评估这些基因在SS的PA患者中的预测准确性。此外,还进行了免疫细胞浸润的广泛分析和单样本基因集富集分析(ssGSEA),以阐明潜在的生物学机制。
使用LIMMA软件包和WGCNA,鉴定出135个与SS中的PA相关的DEG。PPI网络分析揭示了17个候选枢纽基因。三种不同机器学习算法鉴定的基因集的交集突出显示CCL4、CSF1R和MX1为关键DEG。ROC分析和列线图构建证明了它们的高预测准确性(AUC:0.971,95%CI:0.941-1.000)。免疫细胞浸润分析显示这些枢纽基因与失调的免疫细胞之间存在显著正相关。此外,ssGSEA为SS中PA的进展提供了关键的生物学见解。
本研究系统地鉴定了三个有前景的枢纽基因(CCL4、CSF1R和MX1),并开发了一种用于预测SS中PA的列线图。免疫细胞浸润分析表明,失调的免疫细胞显著促进了PA的进展。此外,ssGSEA分析为SS导致PA的机制提供了重要见解。