Lian Ling Zhi, Huang Fang, Lang Jia, Yuan Jing Fang, Hu Ping Ping
Department of Pathology, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200062, China.
Department of Pathology, Zhenjiang Hospital of Chinese Traditional and Western Medicine, Zhenjiang, China.
World J Oncol. 2025 Jul 8;16(4):397-408. doi: 10.14740/wjon2604. eCollection 2025 Aug.
Lung adenocarcinoma (LUAD), the predominant histological subtype of lung cancer, persists in presenting a dismally low 5-year overall survival (OS) rate, notwithstanding advancements in treatment modalities. There exists a pressing necessity for the identification of innovative biomarkers that can enhance prognostic assessments and facilitate individualized therapeutic strategies. The objective of this investigation was to clarify the involvement of genes associated with metabolic reprogramming in the progression of LUAD and to evaluate their viability as prognostic indicators.
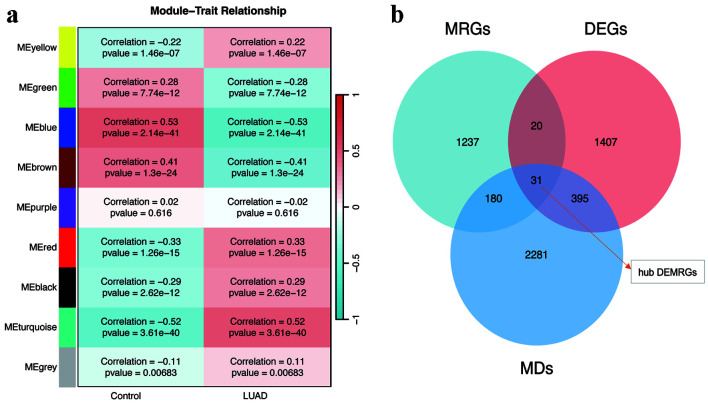
An analysis of differential gene expression was performed utilizing The Cancer Genome Atlas (TCGA)-LUAD dataset, supplemented by a weighted gene co-expression network analysis (WGCNA). Through intersection analysis focusing on metabolic reprogramming genes (MRGs), pivotal differentially expressed metabolic reprogramming genes (hub DEMRGs) were identified. Consensus clustering categorized patients into subtypes based on these genes. Functional enrichment analysis and immune microenvironment characterization were conducted, followed by Cox and least absolute shrinkage and selection operator (LASSO) regression analyses to construct a prognostic risk model.
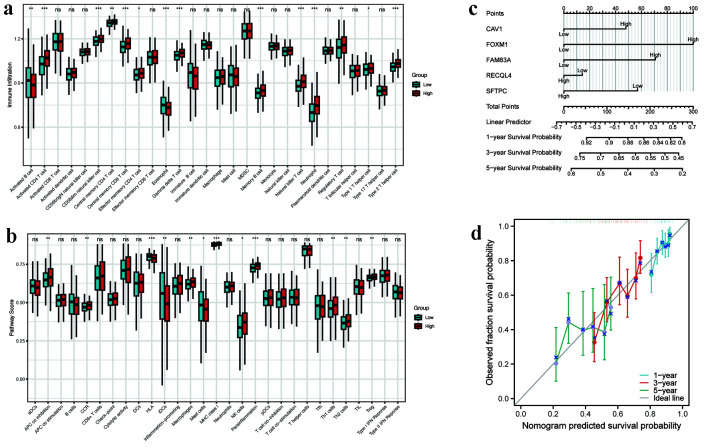
A total of 31 hub DEMRGs were identified. Patients were classified into two distinct subtypes (C1 and C2), with the C2 subtype exhibiting a markedly reduced OS rate. Functional enrichment revealed significant activation of nuclear division and cell cycle pathways in C2. Immune profiling demonstrated an immunosuppressive phenotype in C2, characterized by elevated M2 macrophage infiltration and reduced CD8 T cells. The risk model based on five critical hub DEMRGs showed robust predictive performance (area under the curve (AUC): 0.68 - 0.71), and high-risk patients displayed unique immune cell infiltration patterns.
This research highlights the critical role of MRGs in LUAD prognosis and their potential for clinical application. The identified subtypes and risk model provide insights into tumor heterogeneity and immunosuppressive mechanisms, offering potential targets for individualized therapy.
肺腺癌(LUAD)是肺癌的主要组织学亚型,尽管治疗方式有所进步,但其5年总生存率(OS)仍然低得令人沮丧。迫切需要鉴定能够改善预后评估并促进个体化治疗策略的创新生物标志物。本研究的目的是阐明与代谢重编程相关的基因在LUAD进展中的作用,并评估它们作为预后指标的可行性。
利用癌症基因组图谱(TCGA)-LUAD数据集进行差异基因表达分析,并辅以加权基因共表达网络分析(WGCNA)。通过聚焦代谢重编程基因(MRG)的交集分析,鉴定出关键的差异表达代谢重编程基因(枢纽DEMRG)。基于这些基因的共识聚类将患者分为不同亚型。进行功能富集分析和免疫微环境特征分析,随后进行Cox和最小绝对收缩和选择算子(LASSO)回归分析以构建预后风险模型。
共鉴定出31个枢纽DEMRG。患者被分为两种不同的亚型(C1和C2),C2亚型的OS率明显降低。功能富集显示C2中核分裂和细胞周期途径显著激活。免疫谱分析显示C2具有免疫抑制表型,其特征是M2巨噬细胞浸润增加和CD8 T细胞减少。基于五个关键枢纽DEMRG的风险模型显示出强大的预测性能(曲线下面积(AUC):0.68 - 0.71),高危患者表现出独特的免疫细胞浸润模式。
本研究突出了MRG在LUAD预后中的关键作用及其临床应用潜力。所鉴定的亚型和风险模型为肿瘤异质性和免疫抑制机制提供了见解,为个体化治疗提供了潜在靶点。