He Yuxian, Zhang Han, Zhang Qinggang, Sun Zewei, Sun Xingang, Xia Ling, Zheng Liangrong, Wang Lihong
Heart Center, Department of Cardiovascular Medicine, Zhejiang Provincial People's Hospital (Affiliated People's Hospital, Hangzhou Medical College), Hangzhou, Zhejiang, China.
Department of Cardiology and Atrial Fibrillation Center, School of Medicine, The First Affiliated Hospital, Zhejiang University, Hangzhou, China.
Acta Physiol (Oxf). 2025 Sep;241(9):e70085. doi: 10.1111/apha.70085.
The aim of this study is to determine the possible role of N-methyl-D-aspartate receptor (NMDAR) dysregulation in the ischemic electrical remodeling observed in patients with myocardial infarction (MI) and elucidate the underlying mechanisms.
Human heart tissue was obtained from the border of the infarct and remote zones of patients with ischemic heart disease, and mouse heart tissue was obtained from the peri-infarct zone. NMDAR expression was detected using immunofluorescence (IF) and Western blotting (WB). Spontaneous ventricular arrhythmias (VAs) in mice were detected using electrocardiogram backpacks. Electrical remodeling post-MI was detected using patch clamp recordings, quantitative real-time polymerase chain reactions, IF, and WB. Mechanistic studies were performed using bioinformatic analysis, plasmid and small interfering RNA transfection, lentiviral packaging, and site-directed mutagenesis.
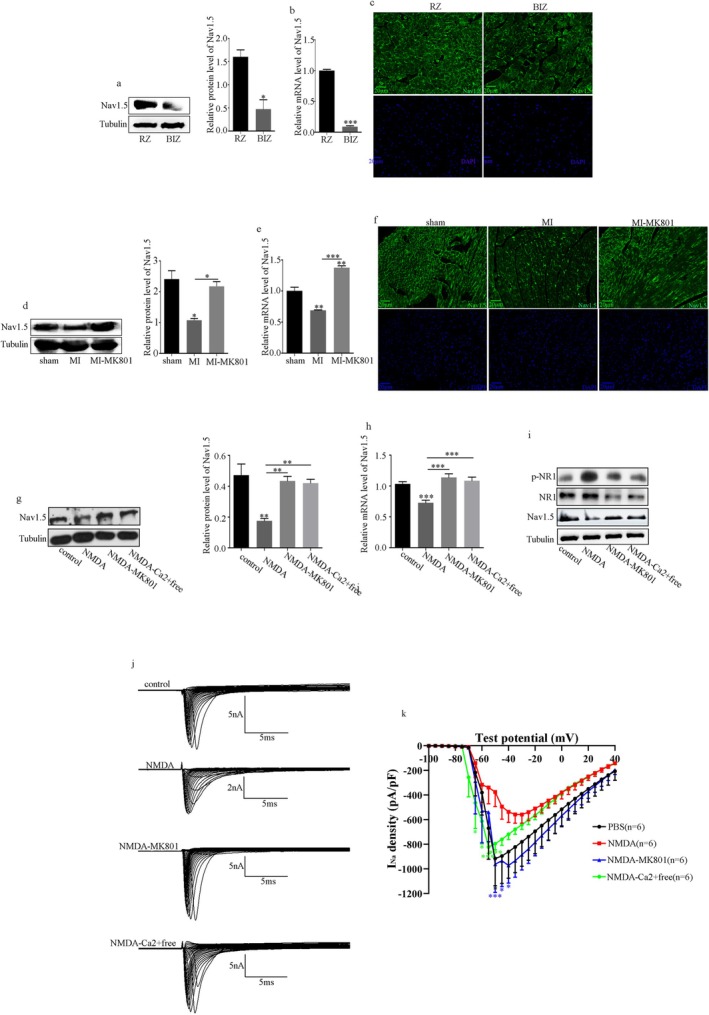
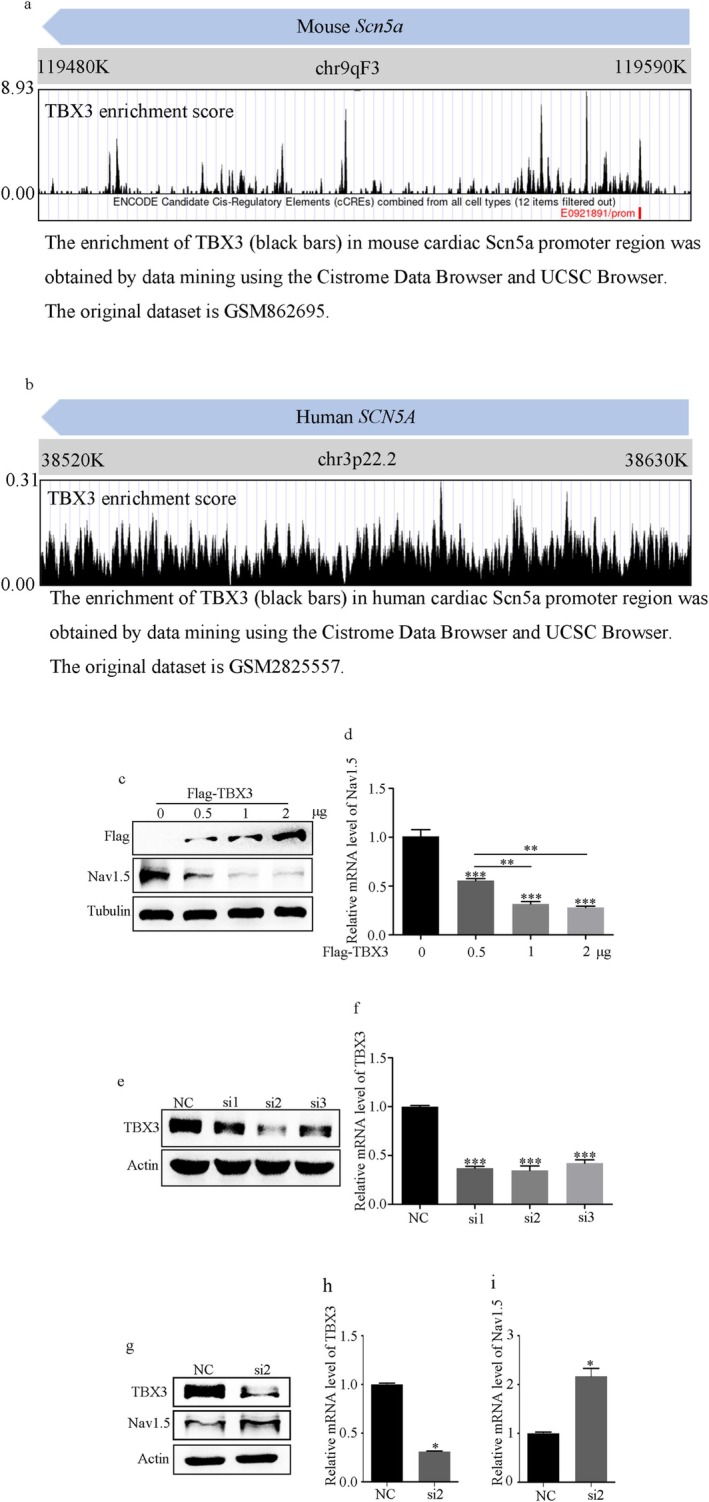
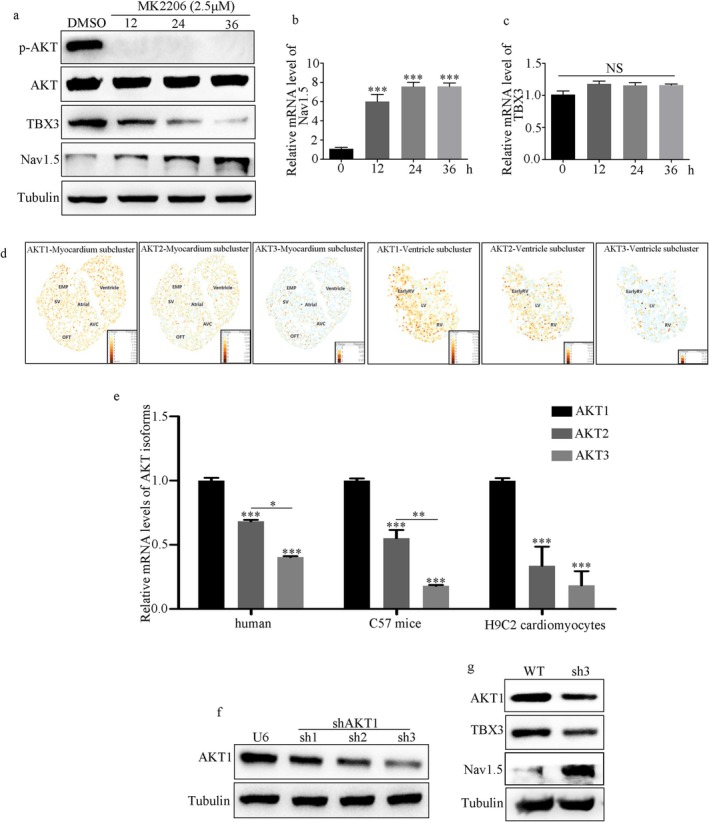
NMDAR is highly expressed in patients with ischemic heart disease and mice with MI. NMDAR inhibition reduces the occurrence of VAs. Mechanistically, NMDAR activation promotes electrophysiological remodeling, as characterized by decreased Nav1.5, Kv11.1, Kv4.2, Kv7.1, Kir2.1, and Cav1.2 expression in patients with ischemic heart disease and mice with MI and rescues these ion channels dysregulation in mice with MI to varying degrees by NMDAR inhibition. Decreased Nav1.5 expression and inward sodium current density were attenuated by NMDAR inhibition in primary rat cardiomyocytes. Moreover, NMDAR activation upregulates T-Box Transcription Factor 3 (TBX3) post-translationally, further downregulating Nav1.5 transcriptionally. Furthermore, AKT1 is the predominant isoform in the ventricular myocardium upstream of TBX3 and mediates NMDAR-induced TBX3 upregulation in cardiomyocytes.
NMDAR activation contributes to MI-induced VAs by regulating the AKT1-TBX3-Nav1.5 axis, providing novel therapeutic strategies for treating ischemic arrhythmias.
本研究旨在确定N-甲基-D-天冬氨酸受体(NMDAR)失调在心肌梗死(MI)患者缺血性电重构中可能发挥的作用,并阐明其潜在机制。
从缺血性心脏病患者的梗死边缘和远隔区域获取人心脏组织,从梗死周边区域获取小鼠心脏组织。采用免疫荧光(IF)和蛋白质免疫印迹法(WB)检测NMDAR表达。使用心电图背包检测小鼠的自发性室性心律失常(VA)。采用膜片钳记录、定量实时聚合酶链反应、IF和WB检测MI后的电重构。通过生物信息学分析、质粒和小干扰RNA转染、慢病毒包装和定点诱变进行机制研究。
NMDAR在缺血性心脏病患者和MI小鼠中高表达。抑制NMDAR可减少VA的发生。机制上,NMDAR激活促进电生理重构,其特征为缺血性心脏病患者和MI小鼠中电压门控钠通道1.5(Nav1.5)、钾通道11.1(Kv11.1)、钾通道4.2(Kv4.2)、钾通道7.1(Kv7.1)、内向整流钾通道2.1(Kir2.1)和L型钙通道α1C亚基(Cav1.2)表达降低,而抑制NMDAR可在不同程度上挽救MI小鼠中这些离子通道的失调。在原代大鼠心肌细胞中,抑制NMDAR可减弱Nav1.5表达降低和内向钠电流密度降低。此外,NMDAR激活在翻译后上调T盒转录因子3(TBX3),进而在转录水平进一步下调Nav1.5。此外,蛋白激酶B1(AKT1)是TBX3上游心室心肌中的主要亚型,介导NMDAR诱导的心肌细胞中TBX3上调。
NMDAR激活通过调节AKT1-TBX3-Nav1.5轴促成MI诱导的VA,为治疗缺血性心律失常提供了新的治疗策略。