Zheng Rui, Yang Wenhao, Yan Jierui, Guo Zhuoyao, Chen Weicheng, Chen Lina, Xu Wenming
Department of Obstetrics/Gynecology, Joint Laboratory of Reproductive Medicine (SCU- CUHK), Key Laboratory of Obstetric, Gynecologic, and Pediatric Diseases and Birth Defects of Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, China.
Department of Pediatric Pulmonology and Immunology, West China Second University Hospital, Sichuan University, Chengdu, China.
Orphanet J Rare Dis. 2025 Sep 2;20(1):469. doi: 10.1186/s13023-025-03977-w.
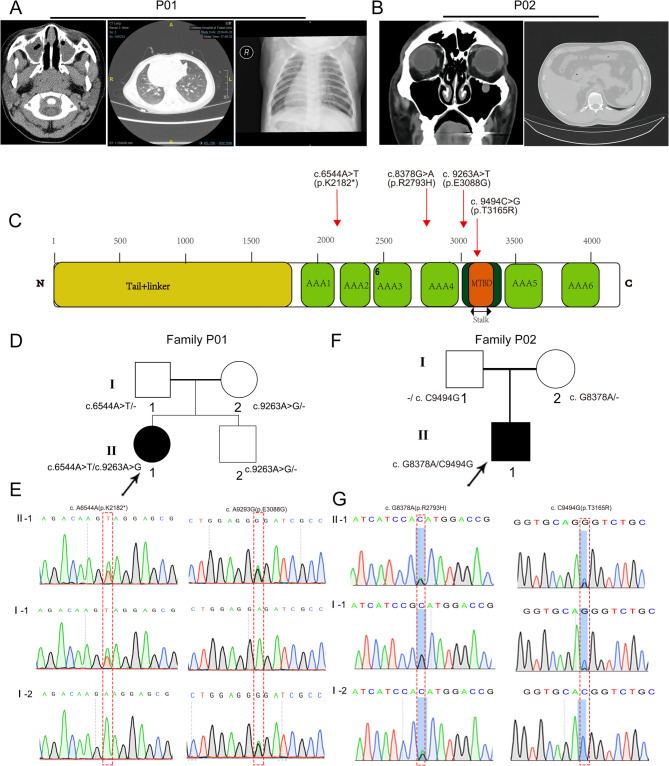
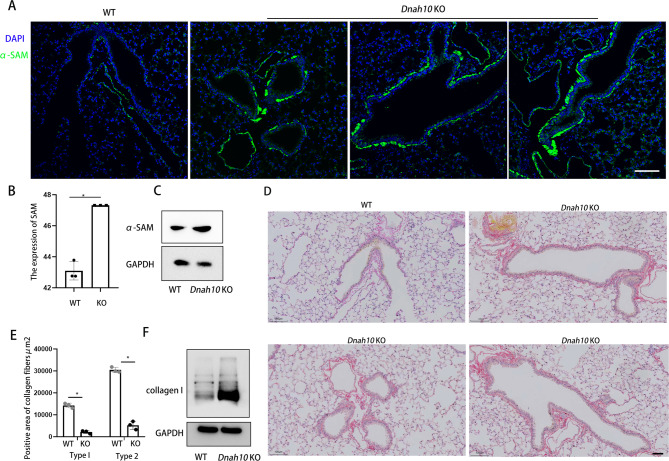
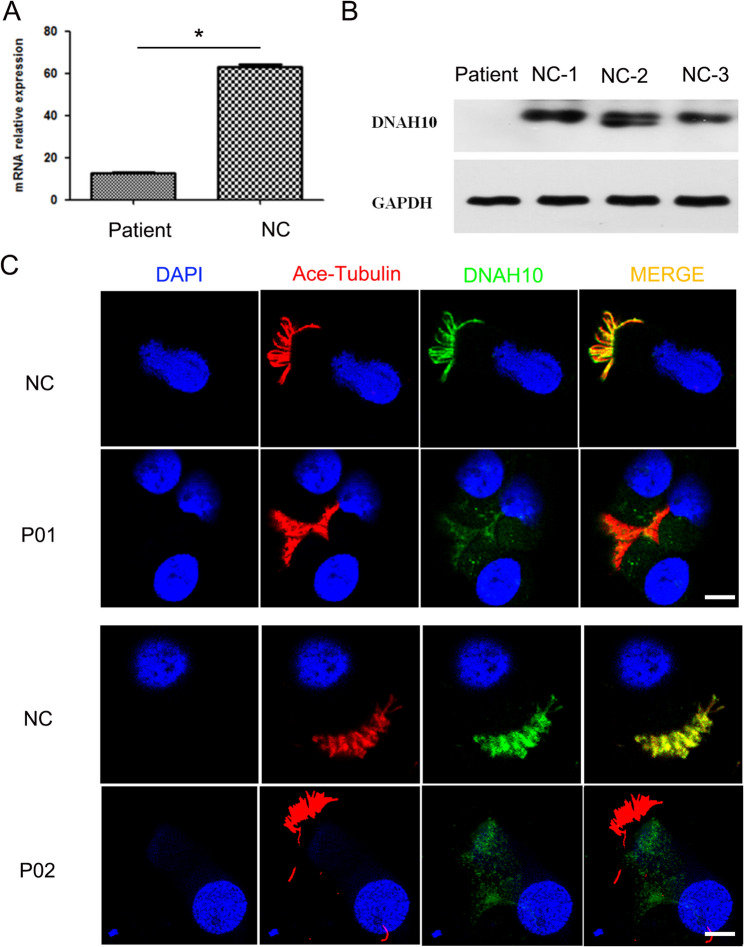
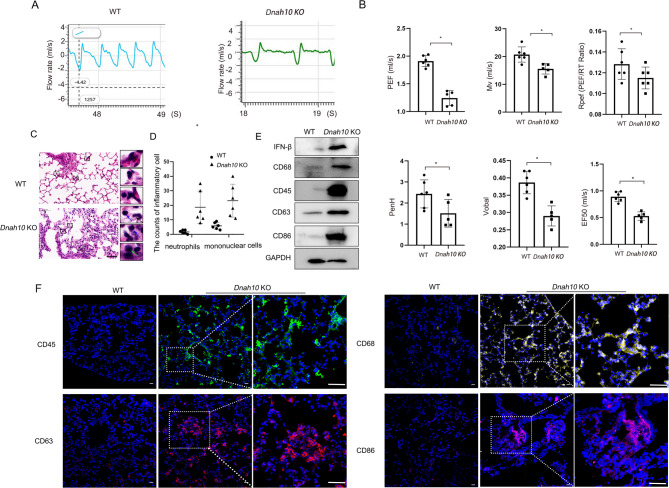
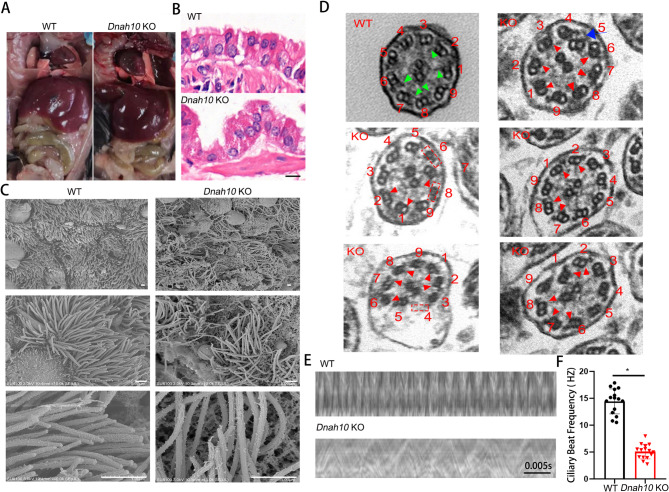
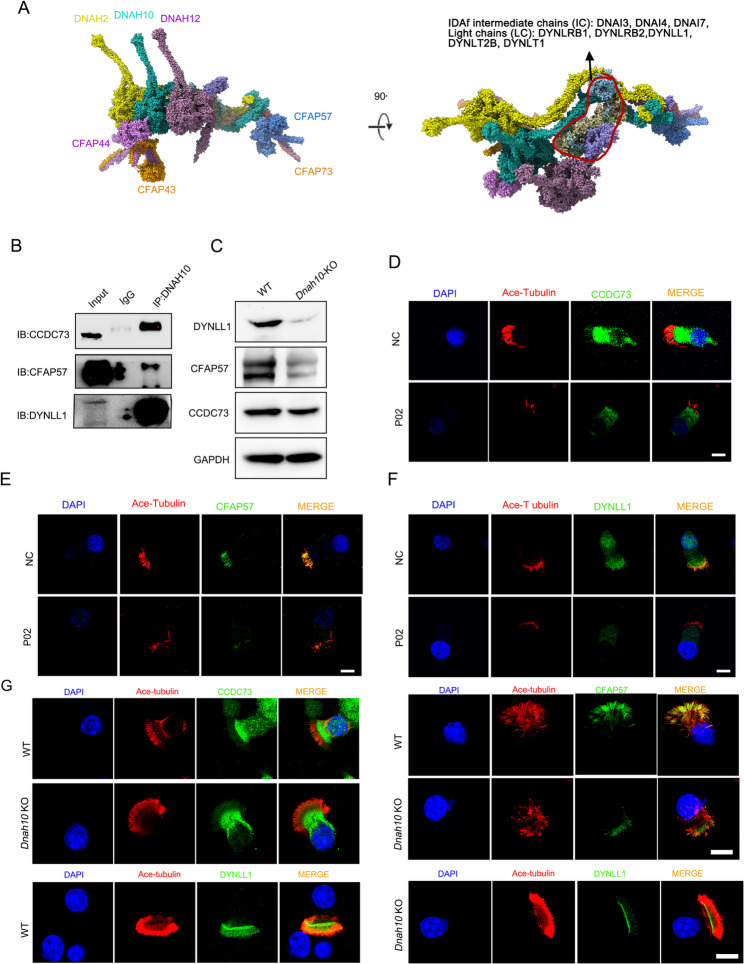
Primary ciliary dyskinesia (PCD; MIM 244400) is a genetic disorder, and its morbidity has been previously underestimated. Mutations in ciliary proteins underlie the disease, resulting in ciliary dysfunction. DNAH10 is an inner arm dynein heavy chain that has been shown to play a critical role in the movement of sperm flagella. In the present study, we demonstrated the presence of loss-of-function mutations in the gene among two families affected by primary ciliary dyskinesia. Patients displayed characteristic symptoms associated with PCD, including chronic respiratory infections, productive cough, and rhinosinusitis. Additionally, a decrease in the expression of DNAH10 was confirmed in both patients. knockout (KO) mice exhibited phenotypic characteristics recapitulating the PCD symptoms observed in two patients. Scanning electron microscope results showed curved and defective cilia morphology in KO mice. Immunostaining also showed that DNAH10 was specifically expressed in the cilia cell. Ciliary structural studies highlighted that DNAH10 interacted with candidate PCD proteins, including CFAP57, DYNLL1, and CCDC73, contributing to the formation of a double-headed inner dynein arm f (IDAf) complex. Co-IP experiment confirmed that DNAH10 can interact with CFAP57, DYNLL1, and CCDC73. We then detected the reduced expression of CFAP57, DYNLL1, and CCDC73 in patient P02 and KO mice. Furthermore, through proteomic analysis, we demonstrated alterations in the expression of abnormal innate immune proteins, super-molecular fiber organization, and mitochondrial respiratory chains. These findings suggested that the loss of DNAH10 leads to improper assembly of the IDAf complex, resulting in ciliary dysfunction and pulmonary fibrosis as the signature manifestation. Notably, our research findings hold substantial implications for the advancement of therapeutic strategies aimed at addressing ciliopathies.
The online version contains supplementary material available at 10.1186/s13023-025-03977-w.
原发性纤毛运动障碍(PCD;MIM 244400)是一种遗传性疾病,其发病率此前一直被低估。纤毛蛋白的突变是该疾病的基础,导致纤毛功能障碍。DNAH10是一种内臂动力蛋白重链,已被证明在精子鞭毛运动中起关键作用。在本研究中,我们在两个受原发性纤毛运动障碍影响的家族中证实了该基因存在功能丧失突变。患者表现出与PCD相关的特征性症状,包括慢性呼吸道感染、咳痰和鼻窦炎。此外,两名患者中均证实DNAH10表达降低。基因敲除(KO)小鼠表现出的表型特征重现了在两名患者中观察到的PCD症状。扫描电子显微镜结果显示KO小鼠的纤毛形态弯曲且有缺陷。免疫染色还显示DNAH10在纤毛细胞中特异性表达。纤毛结构研究突出表明,DNAH10与候选PCD蛋白相互作用,包括CFAP57、DYNLL1和CCDC73,有助于形成双头内动力蛋白臂f(IDAf)复合体。免疫共沉淀实验证实DNAH10可与CFAP57、DYNLL1和CCDC73相互作用。然后我们检测到患者P02和KO小鼠中CFAP57、DYNLL1和CCDC73的表达降低。此外,通过蛋白质组学分析,我们证明了异常固有免疫蛋白、超分子纤维组织和线粒体呼吸链的表达发生了改变。这些发现表明,DNAH10的缺失导致IDAf复合体组装不当,导致纤毛功能障碍和肺纤维化作为标志性表现。值得注意的是,我们的研究结果对旨在治疗纤毛病的治疗策略的推进具有重要意义。
在线版本包含可在10.1186/s13023-025-03977-w获取的补充材料。