Boerkoel C F, Exelbert R, Nicastri C, Nichols R C, Miller F W, Plotz P H, Raben N
Arthritis and Rheumatism Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institute of Health, Bethesda, MD 20892, USA.
Am J Hum Genet. 1995 Apr;56(4):887-97.
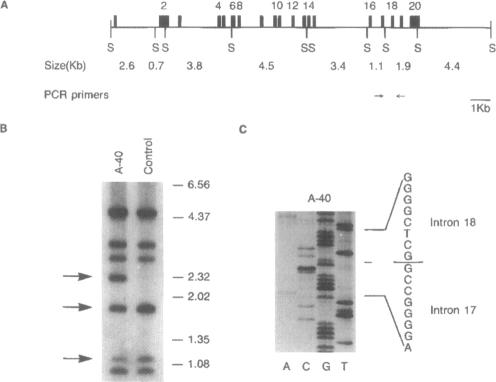
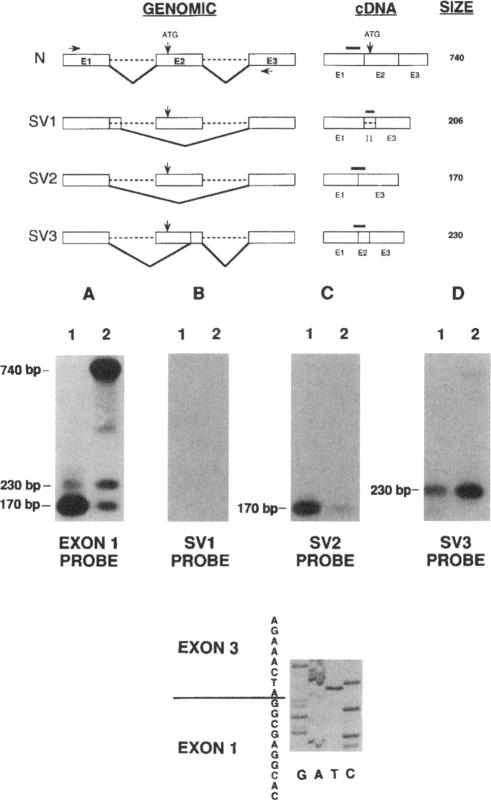
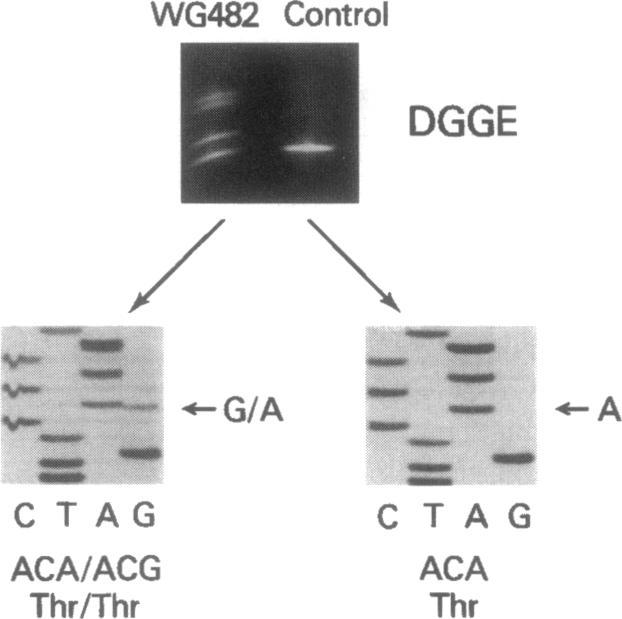
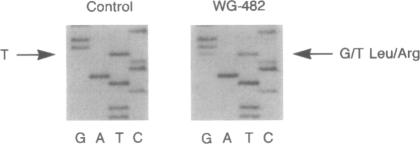
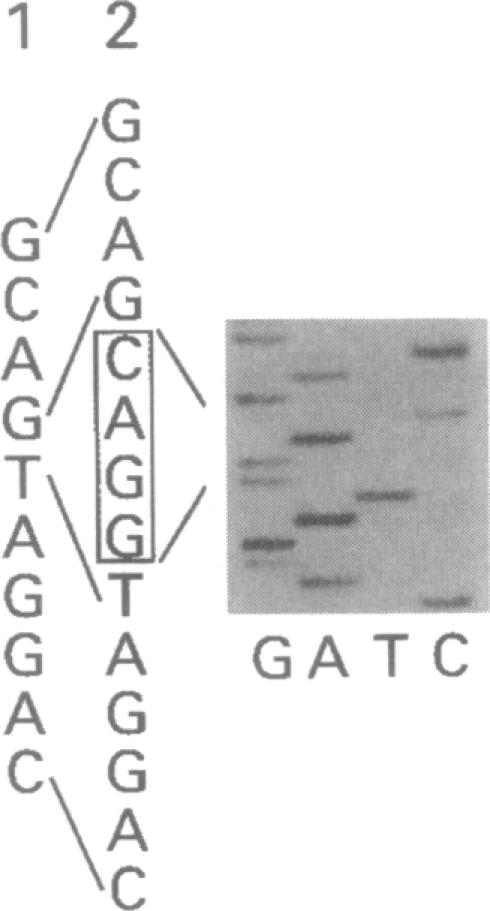
An autosomal recessive deficiency of acid alpha-glucosidase (GAA), type II glycogenosis, is genetically and clinically heterogeneous. The discovery of an enzyme-inactivating genomic deletion of exon 18 in three unrelated genetic compound patients--two infants and an adult--provided a rare opportunity to analyze the effect of the second mutation in patients who displayed dramatically different phenotypes. A deletion of Lys-903 in one patient and a substitution of Arg for Leu-299 in another resulted in the fatal infantile form. In the adult, a T-to-G base change at position -13 of intron 1 resulted in alternatively spliced transcripts with deletion of exon 2, the location of the start codon. The low level of active enzyme (12% of normal) generated from the leakage of normally spliced mRNA sustained the patient to adult life.
酸性α-葡萄糖苷酶(GAA)的常染色体隐性缺乏,即II型糖原贮积病,在遗传和临床方面具有异质性。在三名无亲缘关系的遗传复合患者(两名婴儿和一名成人)中发现了第18外显子的酶失活基因组缺失,这为分析具有显著不同表型的患者中第二个突变的影响提供了难得的机会。一名患者赖氨酸903缺失,另一名患者亮氨酸299被精氨酸替代,导致致命的婴儿型。在成人患者中,内含子1第-13位的T到G碱基变化导致外显子2缺失的可变剪接转录本,外显子2是起始密码子的位置。正常剪接的mRNA泄漏产生的低水平活性酶(正常水平的12%)使该患者活到成年。