Thomas G R, Jensson O, Gudmundsson G, Thorsteinsson L, Cox D W
Research Institute, Hospital for Sick Children, Toronto, Ontario, Canada.
Am J Hum Genet. 1995 May;56(5):1140-6.
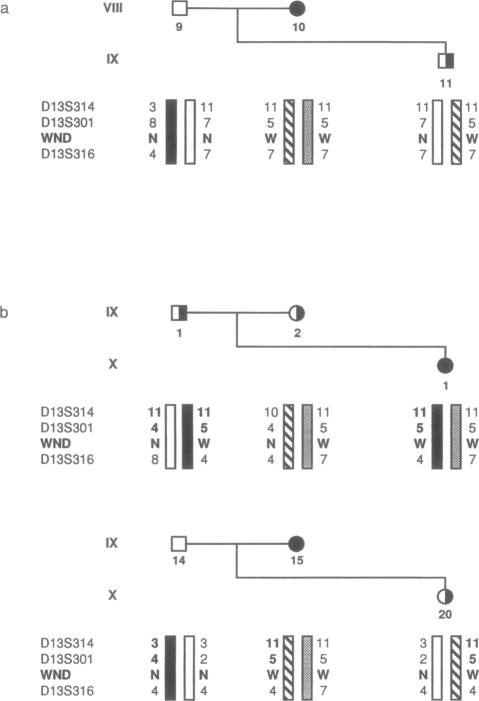
A survey of Wilson disease in Iceland has revealed two large kindreds with affected individuals. We have carried out studies of haplotypes of dinucleotide repeat polymorphisms (CA repeats) flanking the Wilson disease gene. The same mutation, a 7-bp deletion, is present in both families, and the clinical features are similar. The haplotype data and nature of the mutation support the existence of a founder chromosome carrying the mutation. This Icelandic mutation was not found in patients of Irish or Scottish origins, who could share some of the Icelandic ancestral genes. Although the protein function is predicted to be completely abolished by the deletion, predicting early-onset liver disease, we find that the patients present with later-onset neurological and psychiatric symptoms. We show that alternative splicing of the transcript in the region of the deletion could contribute to later onset, suggesting that alternative isoforms of the protein might have some functional significance.
冰岛对威尔逊氏病的一项调查发现了两个有患病个体的大家族。我们对威尔逊氏病基因侧翼的二核苷酸重复多态性(CA重复)单倍型进行了研究。两个家族都存在相同的突变,即一个7碱基对的缺失,且临床特征相似。单倍型数据和突变性质支持存在携带该突变的奠基者染色体。在具有部分冰岛祖先基因的爱尔兰或苏格兰裔患者中未发现这种冰岛突变。尽管预测该缺失会完全消除蛋白质功能,从而预示早发性肝病,但我们发现患者表现为迟发性神经和精神症状。我们表明,缺失区域转录本的可变剪接可能导致发病延迟,这表明该蛋白质的可变异构体可能具有一定的功能意义。