Cao Z, Natowicz M R, Kaback M M, Lim-Steele J S, Prence E M, Brown D, Chabot T, Triggs-Raine B L
Department of Biochemistry and Molecular Biology, University of Manitoba, Winnipeg, Canada.
Am J Hum Genet. 1993 Dec;53(6):1198-205.
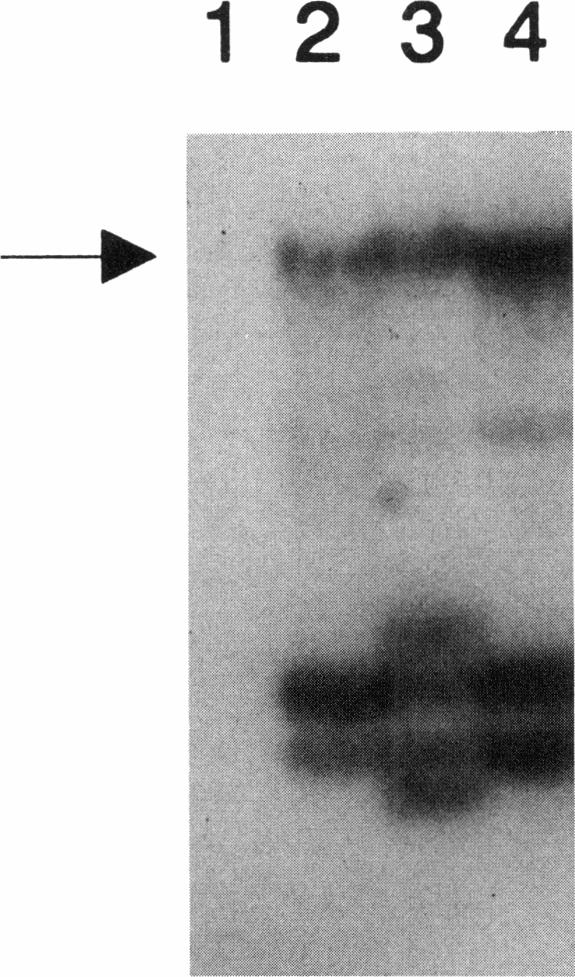
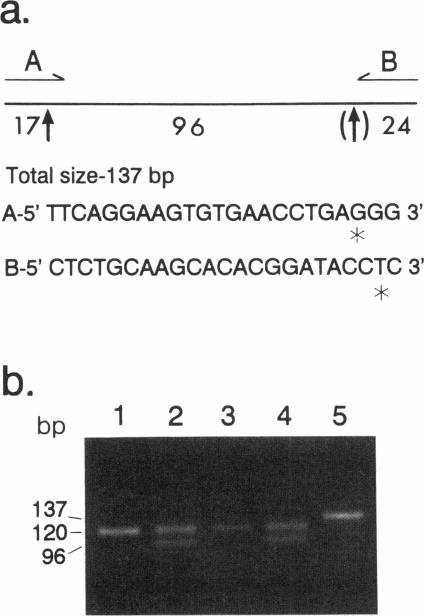
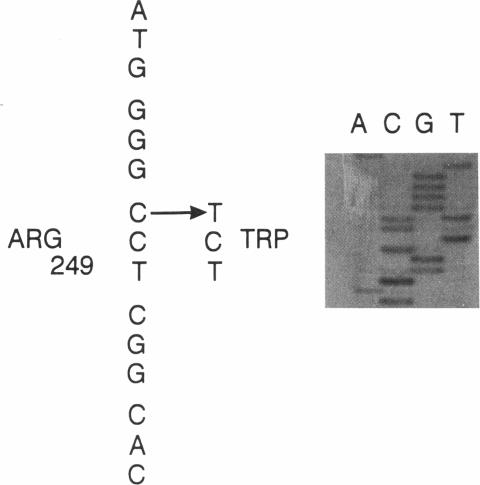
Deficient activity of beta-hexosaminidase A (Hex A), resulting from mutations in the HEXA gene, typically causes Tay-Sachs disease. However, healthy individuals lacking Hex A activity against synthetic substrates (i.e., individuals who are pseudodeficient) have been described. Recently, an apparently benign C739-to-T (Arg247Trp) mutation was found among individuals with Hex A levels indistinguishable from those of carriers of Tay-Sachs disease. This allele, when in compound heterozygosity with a second "disease-causing" allele, results in Hex A pseudodeficiency. We examined the HEXA gene of a healthy 42-year-old who was Hex A deficient but did not have the C739-to-T mutation. The HEXA exons were PCR amplified, and the products were analyzed for mutations by using restriction-enzyme digestion or single-strand gel electrophoresis. A G805-to-A (Gly269Ser) mutation associated with adult-onset GM2 gangliosidosis was found on one chromosome. A new mutation, C745-to-T (Arg249Trp), was identified on the second chromosome. This mutation was detected in an additional 4/63 (6%) non-Jewish and 0/218 Ashkenazi Jewish enzyme-defined carriers. Although the Arg249Trp change may result in a late-onset form of GM2 gangliosidosis, any phenotype must be very mild. This new mutation and the benign C739-to-T mutation together account for approximately 38% of non-Jewish enzyme-defined carriers. Because carriers of the C739-to-T and C745-to-T mutations cannot be differentiated from carriers of disease-causing alleles by using the classical biochemical screening approaches, DNA-based analyses for these mutations should be offered for non-Jewish enzyme-defined heterozygotes, before definitive counseling is provided.
由HEXA基因突变导致的β-己糖胺酶A(Hex A)活性缺乏通常会引起泰-萨克斯病。然而,已经有报道称存在一些健康个体,他们对合成底物缺乏Hex A活性(即假性缺陷个体)。最近,在Hex A水平与泰-萨克斯病携带者无法区分的个体中发现了一种明显良性的C739突变为T(Arg247Trp)的突变。当该等位基因与第二个“致病”等位基因处于复合杂合状态时,会导致Hex A假性缺陷。我们检测了一名42岁健康个体的HEXA基因,该个体Hex A缺乏但没有C739突变为T的突变。对HEXA外显子进行PCR扩增,然后使用限制性酶切或单链凝胶电泳分析产物中的突变。在一条染色体上发现了一个与成人型GM2神经节苷脂病相关的G805突变为A(Gly269Ser)的突变。在第二条染色体上鉴定出一个新的突变,C745突变为T(Arg249Trp)。在另外4/63(6%)的非犹太人和0/218的阿什肯纳兹犹太人酶定义携带者中检测到了这种突变。尽管Arg249Trp的改变可能导致GM2神经节苷脂病的迟发型,但任何表型都必定非常轻微。这个新突变和良性的C739突变为T的突变一起约占非犹太酶定义携带者的38%。由于使用经典生化筛查方法无法将C739突变为T和C745突变为T突变的携带者与致病等位基因的携带者区分开来,因此在为非犹太酶定义的杂合子提供明确咨询之前,应该对这些突变进行基于DNA的分析。