Schoonjans K, Peinado-Onsurbe J, Lefebvre A M, Heyman R A, Briggs M, Deeb S, Staels B, Auwerx J
U325 INSERM, Département d'Athérosclérose, Institut Pasteur, Lille, France.
EMBO J. 1996 Oct 1;15(19):5336-48.
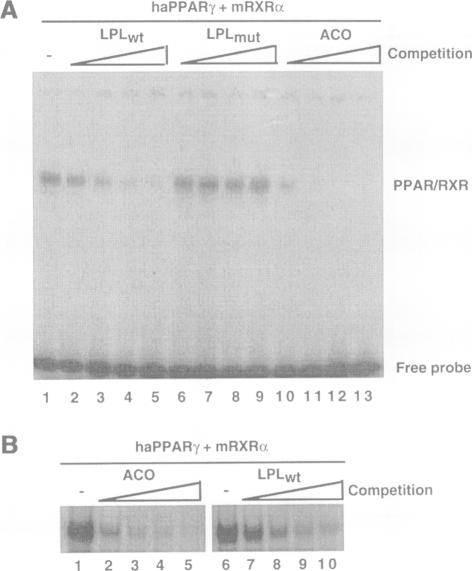
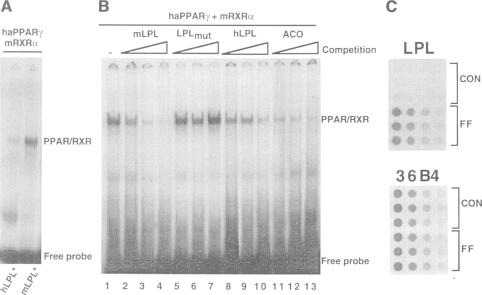
Increased activity of lipoprotein lipase (LPL) may explain the hypotriglyceridemic effects of fibrates, thiazolidinediones and fatty acids, which are known activators (and/or ligands) of the various peroxisome proliferator-activated receptors (PPARs). Treatment with compounds which activate preferentially PPARalpha, such as fenofibrate, induced LPL expression exclusively in rat liver. In contrast, the antidiabetic thiazolidinedione BRL 49653, a high affinity ligand for PPARgamma, had no effect on liver, but induced LPL expression in rat adipose tissue. In the hepatocyte cell line AML-12, fenofibric acid, but not BRL 49653, induced LPL mRNA, whereas in 3T3-L1 preadipocytes, the PPARgamma ligand induced LPL mRNA levels much quicker and to a higher extent than fenofibric acid. In both the in vivo and in vitro studies, inducibility by either PPARalpha or gamma activators, correlated with the tissue distribution of the respective PPARs: an adipocyte-restricted expression of PPARgamma, whereas PPARalpha was expressed predominantly in liver. A sequence element was identified in the human LPL promoter that mediates the functional responsiveness to fibrates and thiazolidinediones. Methylation interference and gel retardation assays demonstrated that a PPARalpha or gamma and the 9-cis retinoic acid receptor (RXR) heterodimers bind to this sequence -169 TGCCCTTTCCCCC -157. These data provide evidence that transcriptional activation of the LPL gene by fibrates and thiazolidinediones is mediated by PPAR-RXR heterodimers and contributes significantly to their hypotriglyceridemic effects in vivo. Whereas thiazolidinediones predominantly affect adipocyte LPL production through activation of PPARgamma, fibrates exert their effects mainly in the liver via activation of PPARalpha.
脂蛋白脂肪酶(LPL)活性增加可能解释了贝特类药物、噻唑烷二酮类药物和脂肪酸的降甘油三酯作用,这些药物是各种过氧化物酶体增殖物激活受体(PPARs)的已知激活剂(和/或配体)。用优先激活PPARα的化合物(如非诺贝特)治疗,仅在大鼠肝脏中诱导LPL表达。相比之下,抗糖尿病噻唑烷二酮类药物BRL 49653是PPARγ的高亲和力配体,对肝脏无影响,但可诱导大鼠脂肪组织中的LPL表达。在肝细胞系AML-12中,非诺贝特酸可诱导LPL mRNA表达,而BRL 49653则不能;而在3T3-L1前脂肪细胞中,PPARγ配体诱导LPL mRNA水平的速度比非诺贝特酸更快,程度更高。在体内和体外研究中,PPARα或γ激活剂的诱导能力与各自PPARs的组织分布相关:PPARγ在脂肪细胞中特异性表达,而PPARα主要在肝脏中表达。在人LPL启动子中鉴定出一个序列元件,该元件介导对贝特类药物和噻唑烷二酮类药物的功能反应性。甲基化干扰和凝胶阻滞试验表明,PPARα或γ与9-顺式视黄酸受体(RXR)异二聚体结合至该序列-169 TGCCCTTTCCCCC -157。这些数据证明,贝特类药物和噻唑烷二酮类药物对LPL基因的转录激活是由PPAR-RXR异二聚体介导的,并且在体内对它们的降甘油三酯作用有显著贡献。噻唑烷二酮类药物主要通过激活PPARγ影响脂肪细胞LPL的产生,而贝特类药物主要通过激活PPARα在肝脏中发挥作用。