Mowat D R, Croaker G D, Cass D T, Kerr B A, Chaitow J, Adès L C, Chia N L, Wilson M J
Department of Clinical Genetics, Royal Alexandra Hospital for Children, Sydney, NSW, Australia.
J Med Genet. 1998 Aug;35(8):617-23. doi: 10.1136/jmg.35.8.617.
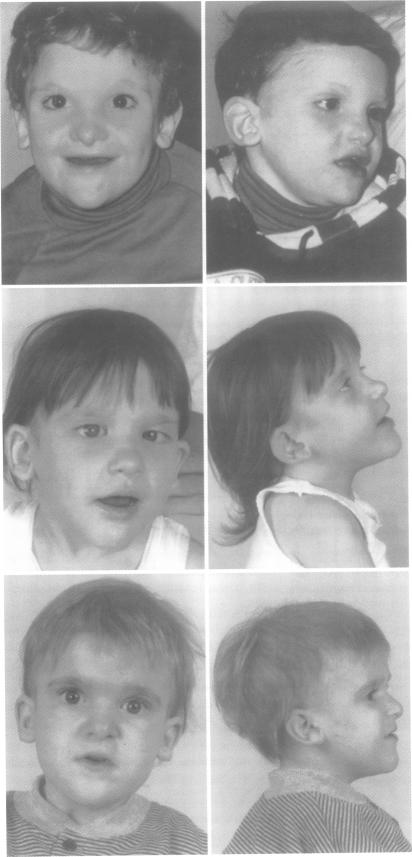
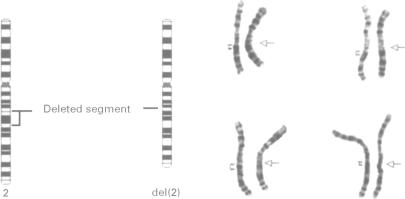
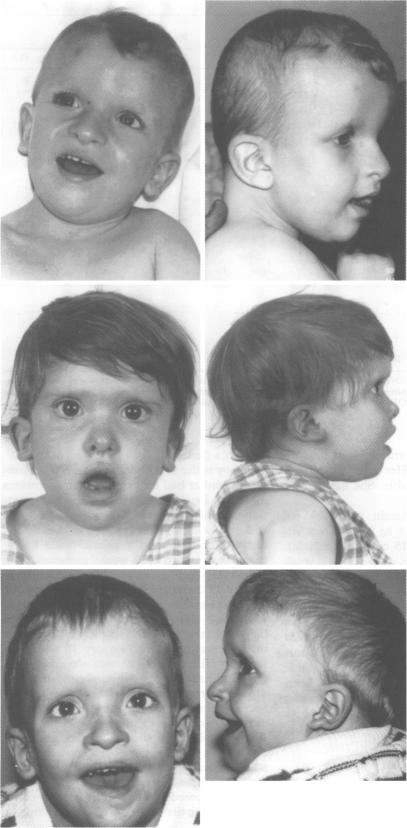
We have identified six children with a distinctive facial phenotype in association with mental retardation (MR), microcephaly, and short stature, four of whom presented with Hirschsprung (HSCR) disease in the neonatal period. HSCR was diagnosed in a further child at the age of 3 years after investigation for severe chronic constipation and another child, identified as sharing the same facial phenotype, had chronic constipation, but did not have HSCR. One of our patients has an interstitial deletion of chromosome 2, del(2)(q21q23). These children strongly resemble the patient reported by Lurie et al with HSCR and dysmorphic features associated with del(2)(q22q23). All patients have been isolated cases, suggesting a contiguous gene syndrome or a dominant single gene disorder involving a locus for HSCR located at 2q22-q23. Review of published reports suggests that there is significant phenotypic and genetic heterogeneity within the group of patients with HSCR, MR, and microcephaly. In particular, our patients appear to have a separate disorder from Goldberg-Shprintzen syndrome, for which autosomal recessive inheritance has been proposed because of sib recurrence and consanguinity in some families.
我们已确定6名患有独特面部表型的儿童,伴有智力发育迟缓(MR)、小头畸形和身材矮小,其中4名儿童在新生儿期患有先天性巨结肠(HSCR)病。在对一名严重慢性便秘患儿进行检查后,于3岁时又诊断出1例HSCR;另一名具有相同面部表型的儿童患有慢性便秘,但未患HSCR。我们的1例患者存在2号染色体间质缺失,del(2)(q21q23)。这些儿童与Lurie等人报道的患有HSCR以及与del(2)(q22q23)相关的畸形特征的患者极为相似。所有患者均为散发病例,提示存在一种涉及位于2q22 - q23的HSCR基因座的连续性基因综合征或显性单基因疾病。对已发表报告的回顾表明,在患有HSCR、MR和小头畸形的患者群体中存在显著的表型和基因异质性。特别是,我们的患者似乎患有与戈德堡 - 施普林岑综合征不同的疾病,由于某些家族中的同胞复发和近亲结婚,有人提出戈德堡 - 施普林岑综合征为常染色体隐性遗传。