Pascual J M, Karlin A
Center for Molecular Recognition, Columbia University, New York 10032, USA.
J Gen Physiol. 1998 Nov;112(5):611-21. doi: 10.1085/jgp.112.5.611.
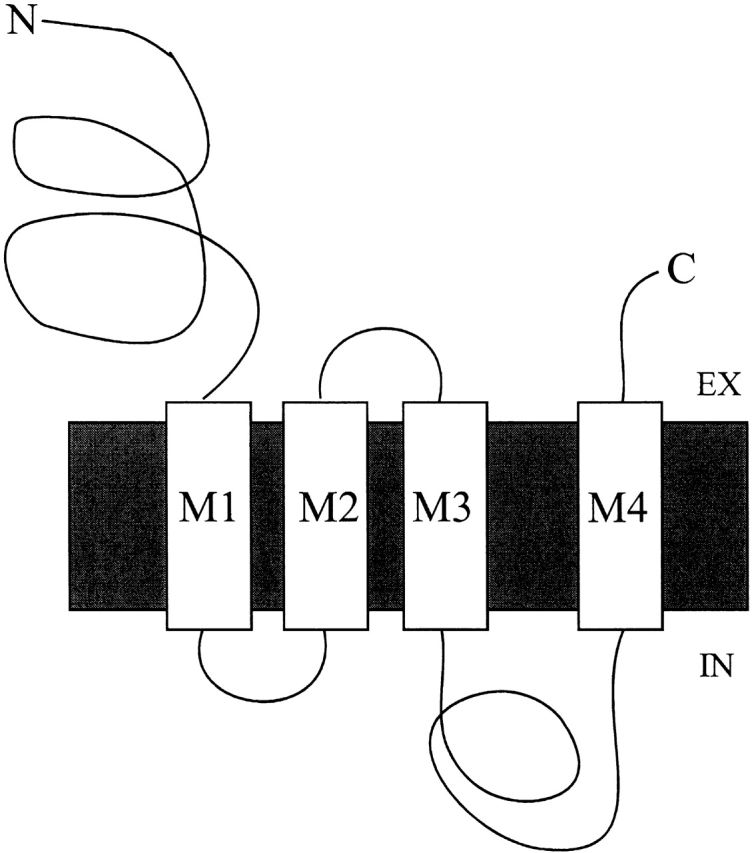
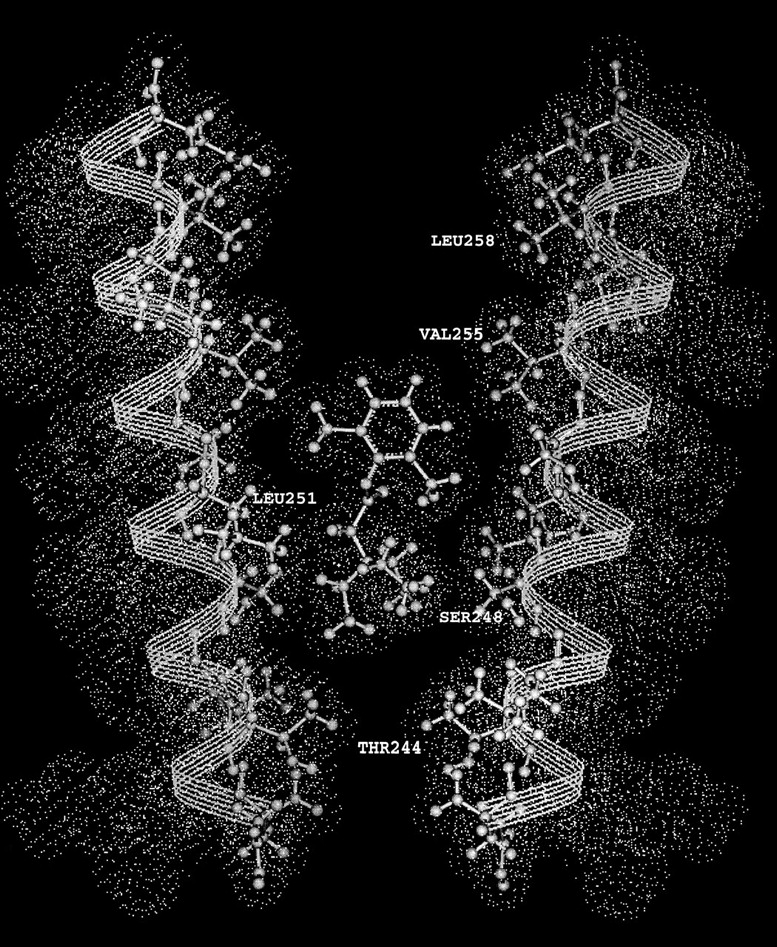
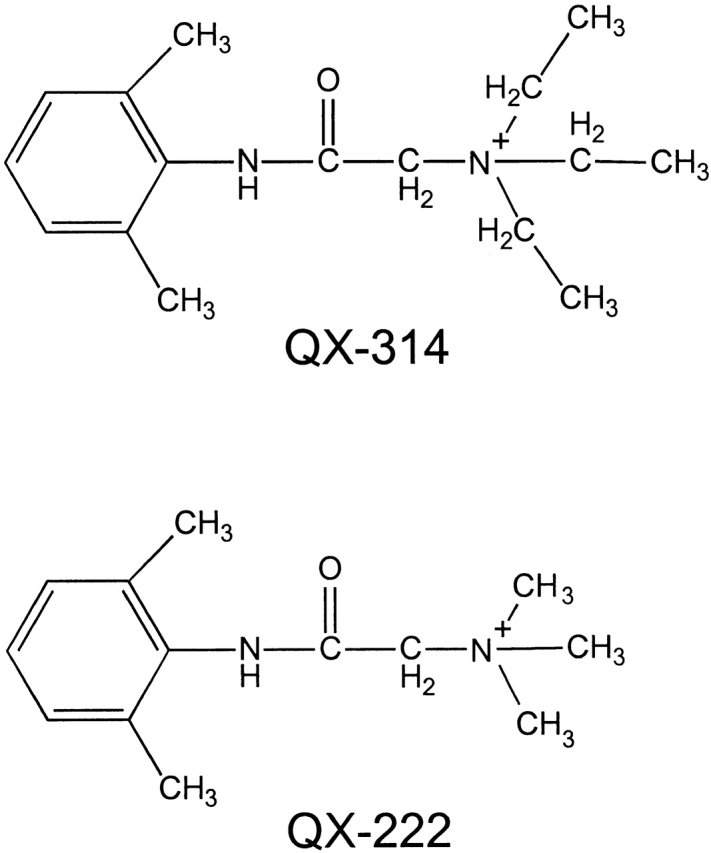
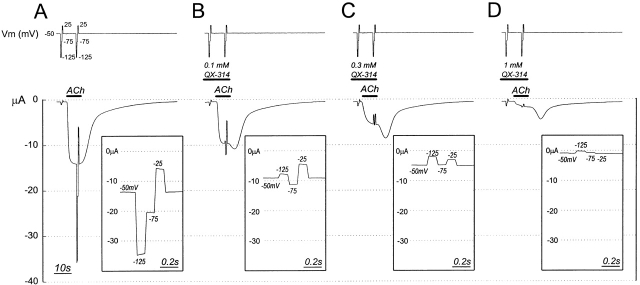
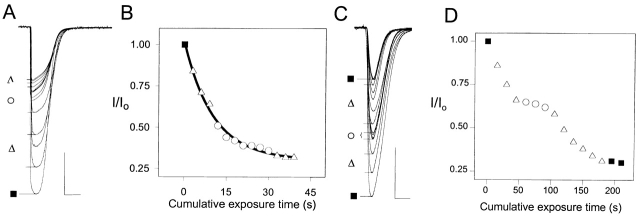
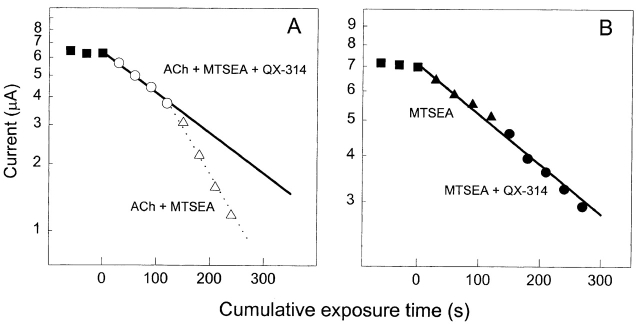
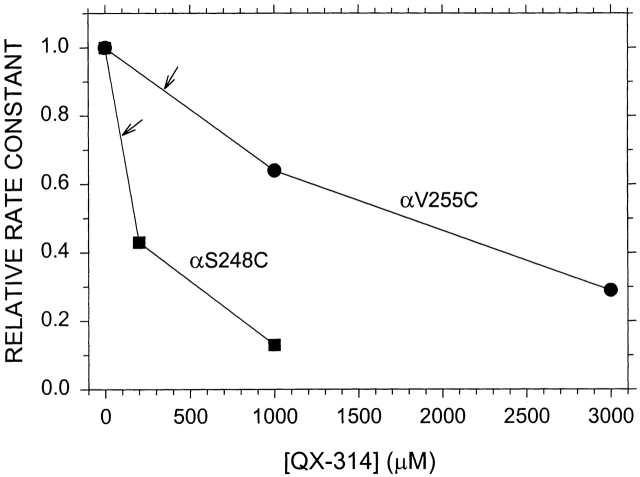
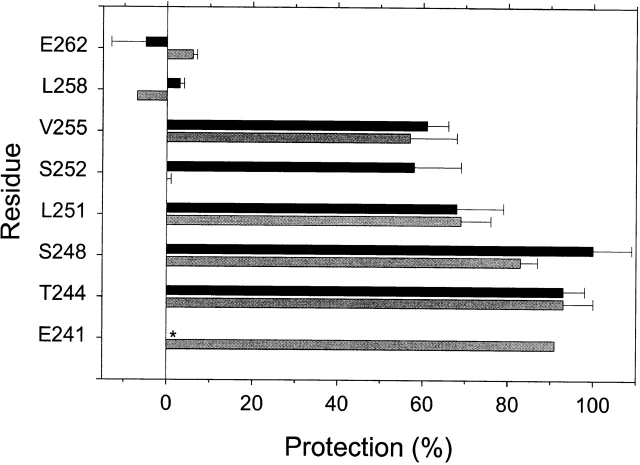
The triethylammonium QX-314 and the trimethylammonium QX-222 are lidocaine derivatives that act as open-channel blockers of the acetylcholine (ACh) receptor. When bound, these blockers should occlude some of the residues lining the channel. Eight residues in the second membrane-spanning segment (M2) of the mouse-muscle alpha subunit were mutated one at a time to cysteine and expressed together with wild-type beta, gamma, and delta subunits in Xenopus oocytes. The rate constant for the reaction of each substituted cysteine with 2-aminoethyl methanethiosulfonate (MTSEA) was determined from the time course of the irreversible effect of MTSEA on the ACh-induced current. The reactions were carried out in the presence and absence of ACh and in the presence and absence of QX-314 and QX-222. These blockers had no effect on the reactions in the absence of ACh. In the presence of ACh, both blockers retarded the reaction of extracellularly applied MTSEA with cysteine substituted for residues from alphaVal255, one third of the distance in from the extracellular end of M2, to alphaGlu241, flanking the intracellular end of M2, but not with cysteine substituted for alphaLeu258 or alphaGlu262, at the extracellular end of M2. The reactions of MTSEA with cysteines substituted for alphaLeu258 and alphaGlu262 were considerably faster in the presence of ACh than in its absence. That QX-314 and QX-222 did not protect alphaL258C and alphaE262C against reaction with MTSEA in the presence of ACh implies that protection of the other residues was due to occlusion of the channel and not to the promotion of a less reactive state from a remote site. Given the 12-A overall length of the blockers and the alpha-helical conformation of M2 in the open state, the binding site for both blockers extends from alphaVal255 down to alphaSer248.
三乙铵QX - 314和三甲铵QX - 222是利多卡因衍生物,可作为乙酰胆碱(ACh)受体的开放通道阻滞剂。结合时,这些阻滞剂应会阻塞通道内衬的一些残基。将小鼠肌肉α亚基第二跨膜片段(M2)中的八个残基一次一个地突变为半胱氨酸,并与野生型β、γ和δ亚基一起在非洲爪蟾卵母细胞中表达。从MTSEA对ACh诱导电流的不可逆作用的时间进程中,测定每个取代半胱氨酸与2 - 氨基乙基甲硫代磺酸盐(MTSEA)反应的速率常数。反应在有和没有ACh的情况下以及有和没有QX - 314和QX - 222的情况下进行。这些阻滞剂在没有ACh时对反应没有影响。在有ACh的情况下,两种阻滞剂都减缓了细胞外应用的MTSEA与取代α亚基Val255(从M₂细胞外端向内三分之一距离处)至α亚基Glu241(M₂细胞内端侧翼)残基的半胱氨酸的反应,但不影响与取代α亚基Leu258或α亚基Glu262(M₂细胞外端)的半胱氨酸的反应。在有ACh的情况下,MTSEA与取代α亚基Leu258和α亚基Glu262的半胱氨酸的反应比没有ACh时快得多。在有ACh的情况下,QX - 314和QX - 222不能保护αL258C和αE262C免受MTSEA的反应,这意味着对其他残基的保护是由于通道的阻塞,而不是由于从远程位点促进反应性较低的状态。鉴于阻滞剂的总长度为12 Å以及开放状态下M₂的α螺旋构象,两种阻滞剂的结合位点从α亚基Val255延伸至α亚基Ser248。