Samudrala Ram, Levitt Michael
Department of Microbiology, University of Washington, School of Medicine, Seattle, WA 98195, USA.
BMC Struct Biol. 2002 Aug 1;2:3. doi: 10.1186/1472-6807-2-3.
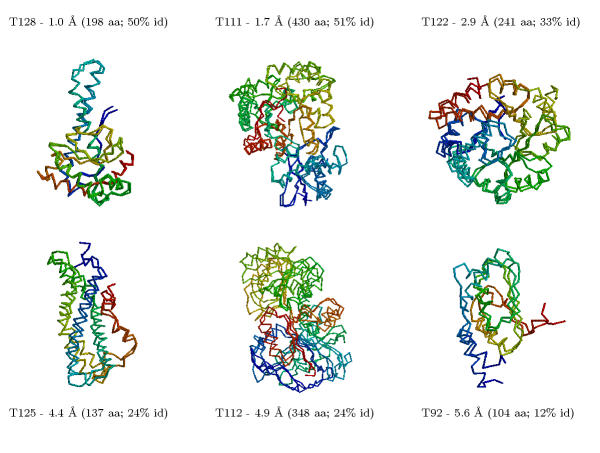
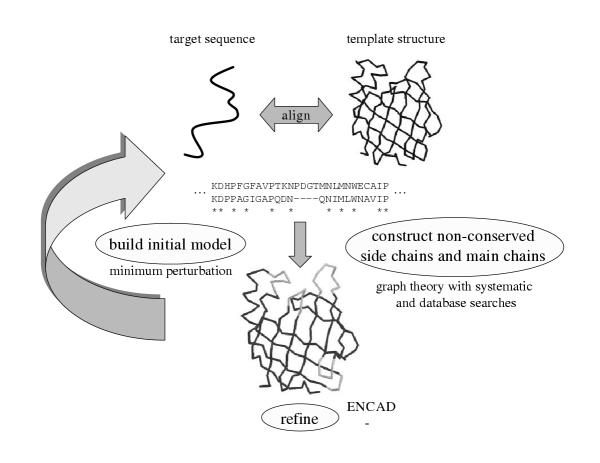
We thoroughly analyse the results of 40 blind predictions for which an experimental answer was made available at the fourth meeting on the critical assessment of protein structure methods (CASP4). Using our comparative modelling and fold recognition methodologies, we made 29 predictions for targets that had sequence identities ranging from 50% to 10% to the nearest related protein with known structure. Using our ab initio methodologies, we made eleven predictions for targets that had no detectable sequence relationships.
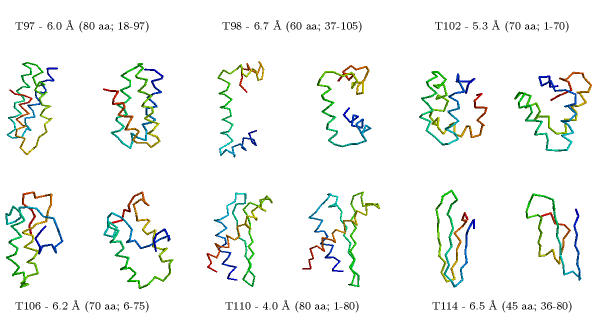
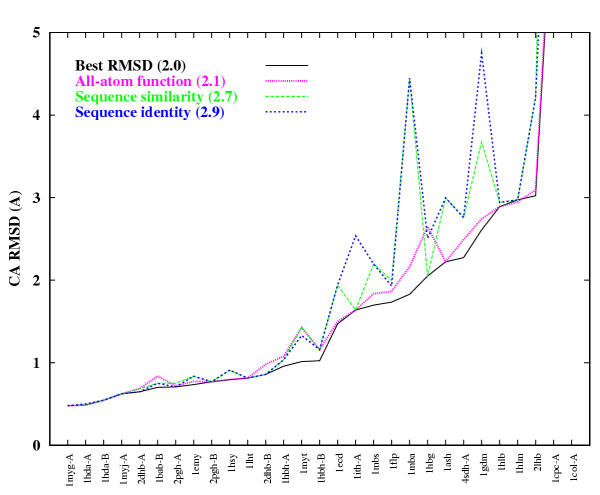
For 23 of these proteins, we produced models ranging from 1.0 to 6.0 A root mean square deviation (RMSD) for the Calpha atoms between the model and the corresponding experimental structure for all or large parts of the protein, with model accuracies scaling fairly linearly with respect to sequence identity (i.e., the higher the sequence identity, the better the prediction). We produced nine models with accuracies ranging from 4.0 to 6.0 A Calpha RMSD for 60-100 residue proteins (or large fragments of a protein), with a prediction accuracy of 4.0 A Calpha RMSD for residues 1-80 for T110/rbfa.
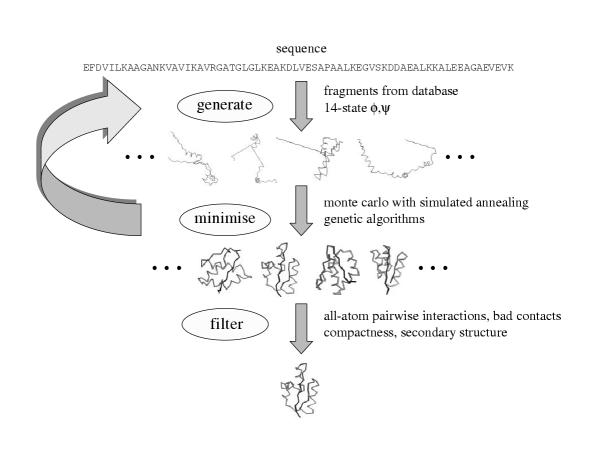
The areas of protein structure prediction that work well, and areas that need improvement, are discernable by examining how our methods have performed over the past four CASP experiments. These results have implications for modelling the structure of all tractable proteins encoded by the genome of an organism.
我们全面分析了在蛋白质结构预测方法关键评估第四次会议(CASP4)上可获得实验答案的40次盲预测结果。使用我们的比较建模和折叠识别方法,我们对与已知结构的最接近相关蛋白质的序列同一性在50%至10%之间的目标进行了29次预测。使用我们的从头算方法,我们对没有可检测到序列关系的目标进行了11次预测。
对于其中23种蛋白质,我们生成的模型在蛋白质的全部或大部分区域中,模型与相应实验结构之间的Cα原子的均方根偏差(RMSD)范围为1.0至6.0埃,模型准确性与序列同一性大致呈线性比例关系(即,序列同一性越高,预测越好)。对于60 - 100个残基的蛋白质(或蛋白质的大片段),我们生成了9个准确性范围为4.0至6.0埃Cα RMSD的模型,对于T110/rbfa的1 - 80位残基,预测准确性为4.0埃Cα RMSD。
通过检查我们的方法在过去四次CASP实验中的表现,可以辨别出蛋白质结构预测中表现良好的领域和需要改进的领域。这些结果对于模拟生物体基因组编码的所有可处理蛋白质的结构具有启示意义。