Pangalos C, Théophile D, Sinet P M, Marks A, Stamboulieh-Abazis D, Chettouh Z, Prieur M, Verellen C, Rethoré M O, Lejeune J
Institut de Progénèse, Paris, France.
Am J Hum Genet. 1992 Dec;51(6):1240-50.
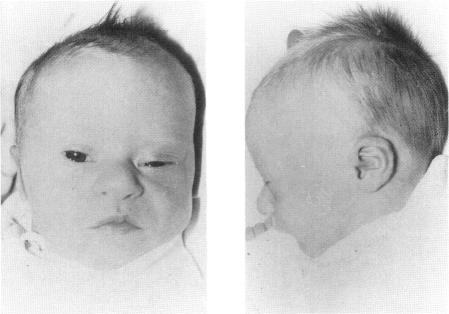
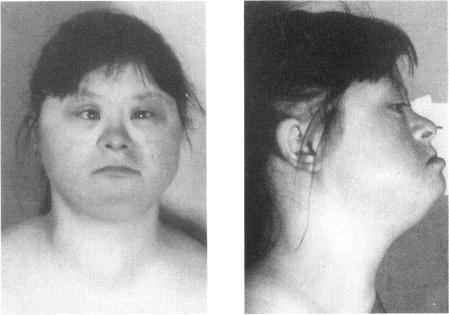
Three Down syndrome patients for whom karyotypic analysis showed a "mirror" (reverse tandem) duplication of chromosome 21 were studied by phenotypic, cytogenetic, and molecular methods. On high-resolution R-banding analysis performed in two cases, the size of the fusion 21q22.3 band was apparently less than twice the size of the normal 21q22.3, suggesting a partial deletion of distal 21q. The evaluation of eight chromosome 21 single-copy sequences of the 21q22 region--namely, SOD1, D21S15, D21S42, CRYA1, PFKL, CD18, COL6A1, and S100B--by a slot blot method showed in all three cases a partial deletion of 21q22.3 and partial monosomy. The translocation breakpoints were different in each patient, and in two cases the rearranged chromosome was found to be asymmetrical. The molecular definition of the monosomy 21 in each patient was, respectively, COL6A1-S100B, CD18-S100B, and PFKL-S100B. DNA polymorphism analysis indicated in all cases a homozygosity of the duplicated material. The duplicated region was maternal in two patients and paternal in one patient. These data suggest that the reverse tandem chromosomes did not result from a telomeric fusion between chromosomes 21 but from a translocation between sister chromatids. The phenotypes of these patients did not differ significantly from that of individuals with full trisomy 21, except in one case with large ears with an unfolded helix. The fact that monosomy of distal 21q22.3 in these patients resulted in a phenotype very similar to Down syndrome suggests that the duplication of the genes located in this part of chromosome 21 is not necessary for the pathogenesis of the Down syndrome features observed in these patients, including most of the facial and hand features, muscular hypotonia, cardiopathy of the Fallot tetralogy type, and part of the mental retardation.
对3例经核型分析显示21号染色体存在“镜像”(反向串联)重复的唐氏综合征患者进行了表型、细胞遗传学和分子学方法研究。在2例患者中进行的高分辨率R带分析显示,融合的21q22.3带的大小明显小于正常21q22.3带大小的两倍,提示21q远端部分缺失。通过狭缝印迹法对21q22区域的8个21号染色体单拷贝序列(即SOD1、D21S15、D21S42、CRYA1、PFKL、CD18、COL6A1和S100B)进行评估,结果显示所有3例患者均存在21q22.3部分缺失和部分单体性。每位患者的易位断点不同,且在2例患者中发现重排染色体不对称。每位患者21号染色体单体性的分子定义分别为COL6A1-S100B、CD18-S100B和PFKL-S100B。DNA多态性分析显示,所有病例中重复物质均为纯合子。重复区域在2例患者中为母源,在1例患者中为父源。这些数据表明,反向串联染色体并非由21号染色体之间的端粒融合产生,而是由姐妹染色单体之间的易位所致。这些患者的表型与21号染色体完全三体的个体相比无显著差异,仅1例患者有耳朵大且耳轮未展开的情况除外。这些患者21q22.3远端单体性导致的表型与唐氏综合征非常相似,这表明位于21号染色体该区域的基因重复对于这些患者所观察到的唐氏综合征特征(包括大多数面部和手部特征、肌张力低下、法洛四联症型心脏病以及部分智力发育迟缓)的发病机制并非必要。