Alexandrov Vadim, Gerstein Mark
Department of Molecular Biophysics and Biochemistry, Yale University, 266 Whitney Ave, New Haven, CT 06511, USA.
BMC Bioinformatics. 2004 Jan 9;5:2. doi: 10.1186/1471-2105-5-2.
Hidden Markov Models (HMMs) have proven very useful in computational biology for such applications as sequence pattern matching, gene-finding, and structure prediction. Thus far, however, they have been confined to representing 1D sequence (or the aspects of structure that could be represented by character strings).
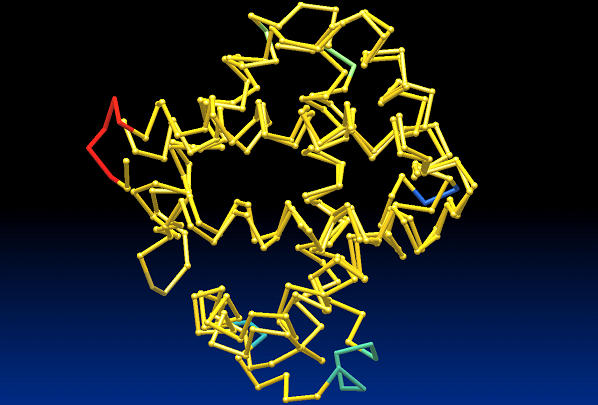
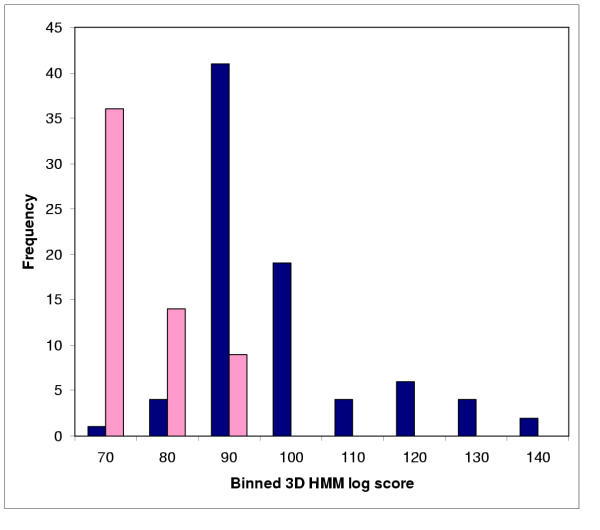
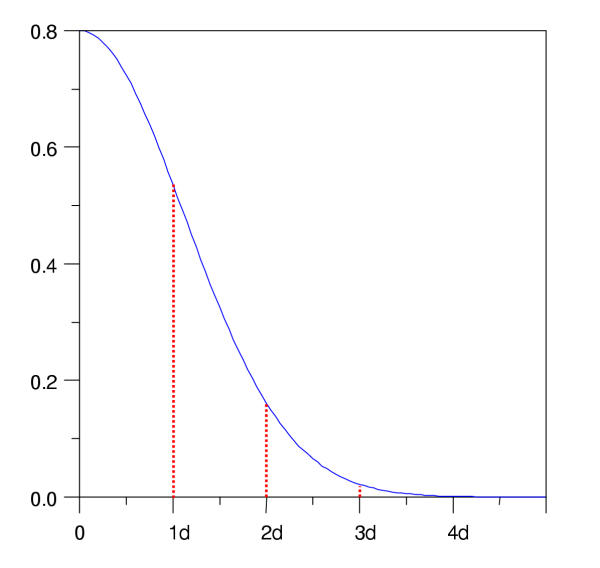
We develop an HMM formalism that explicitly uses 3D coordinates in its match states. The match states are modeled by 3D Gaussian distributions centered on the mean coordinate position of each alpha carbon in a large structural alignment. The transition probabilities depend on the spread of the neighboring match states and on the number of gaps found in the structural alignment. We also develop methods for aligning query structures against 3D HMMs and scoring the result probabilistically. For 1D HMMs these tasks are accomplished by the Viterbi and forward algorithms. However, these will not work in unmodified form for the 3D problem, due to non-local quality of structural alignment, so we develop extensions of these algorithms for the 3D case. Several applications of 3D HMMs for protein structure classification are reported. A good separation of scores for different fold families suggests that the described construct is quite useful for protein structure analysis.
We have created a rigorous 3D HMM representation for protein structures and implemented a complete set of routines for building 3D HMMs in C and Perl. The code is freely available from http://www.molmovdb.org/geometry/3dHMM, and at this site we also have a simple prototype server to demonstrate the features of the described approach.
隐马尔可夫模型(HMM)在计算生物学中已被证明在序列模式匹配、基因查找和结构预测等应用中非常有用。然而,到目前为止,它们仅限于表示一维序列(或可以用字符串表示的结构方面)。
我们开发了一种HMM形式体系,在其匹配状态中明确使用三维坐标。匹配状态由以大型结构比对中每个α碳原子的平均坐标位置为中心的三维高斯分布建模。转移概率取决于相邻匹配状态的分布以及在结构比对中发现的间隙数量。我们还开发了将查询结构与三维HMM进行比对并对结果进行概率评分的方法。对于一维HMM,这些任务由维特比算法和前向算法完成。然而,由于结构比对的非局部性质,这些算法未经修改不能用于三维问题,因此我们针对三维情况开发了这些算法的扩展。报告了三维HMM在蛋白质结构分类中的几个应用。不同折叠家族得分的良好分离表明所描述的构建体对蛋白质结构分析非常有用。
我们为蛋白质结构创建了一种严格的三维HMM表示,并在C和Perl中实现了一套完整的构建三维HMM的例程。代码可从http://www.molmovdb.org/geometry/3dHMM免费获取,在该网站我们还有一个简单的原型服务器来展示所描述方法的功能。