Bagos Pantelis G, Liakopoulos Theodore D, Spyropoulos Ioannis C, Hamodrakas Stavros J
Department of Cell Biology and Biophysics, Faculty of Biology, University of Athens, Panepistimiopolis, Athens 15701, GREECE.
BMC Bioinformatics. 2004 Mar 15;5:29. doi: 10.1186/1471-2105-5-29.
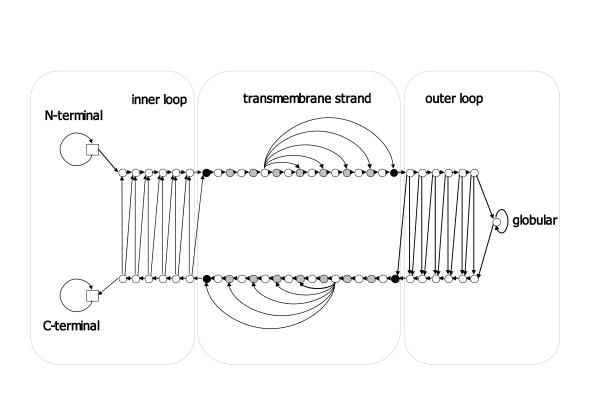
Integral membrane proteins constitute about 20-30% of all proteins in the fully sequenced genomes. They come in two structural classes, the alpha-helical and the beta-barrel membrane proteins, demonstrating different physicochemical characteristics, structure and localization. While transmembrane segment prediction for the alpha-helical integral membrane proteins appears to be an easy task nowadays, the same is much more difficult for the beta-barrel membrane proteins. We developed a method, based on a Hidden Markov Model, capable of predicting the transmembrane beta-strands of the outer membrane proteins of gram-negative bacteria, and discriminating those from water-soluble proteins in large datasets. The model is trained in a discriminative manner, aiming at maximizing the probability of correct predictions rather than the likelihood of the sequences.
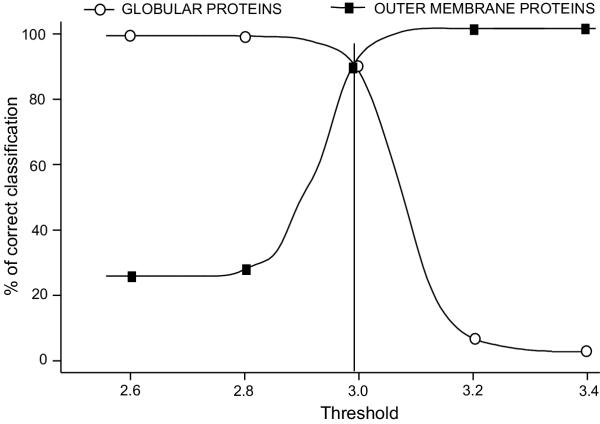
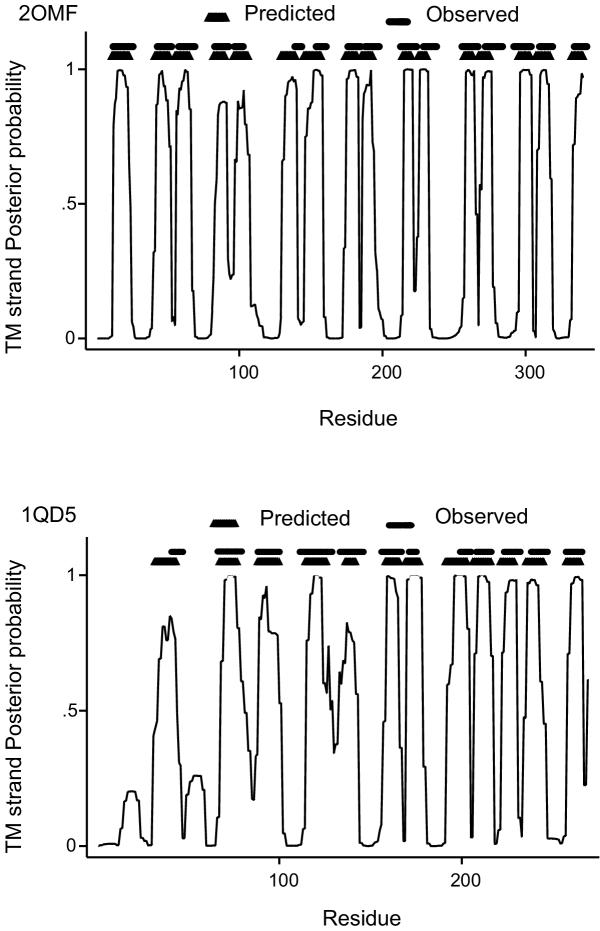
The training has been performed on a non-redundant database of 14 outer membrane proteins with structures known at atomic resolution; it has been tested with a jacknife procedure, yielding a per residue accuracy of 84.2% and a correlation coefficient of 0.72, whereas for the self-consistency test the per residue accuracy was 88.1% and the correlation coefficient 0.824. The total number of correctly predicted topologies is 10 out of 14 in the self-consistency test, and 9 out of 14 in the jacknife. Furthermore, the model is capable of discriminating outer membrane from water-soluble proteins in large-scale applications, with a success rate of 88.8% and 89.2% for the correct classification of outer membrane and water-soluble proteins respectively, the highest rates obtained in the literature. That test has been performed independently on a set of known outer membrane proteins with low sequence identity with each other and also with the proteins of the training set.
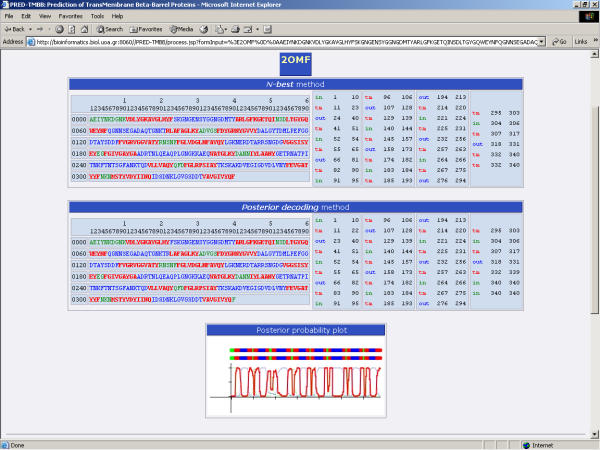
Based on the above, we developed a strategy, that enabled us to screen the entire proteome of E. coli for outer membrane proteins. The results were satisfactory, thus the method presented here appears to be suitable for screening entire proteomes for the discovery of novel outer membrane proteins. A web interface available for non-commercial users is located at: http://bioinformatics.biol.uoa.gr/PRED-TMBB, and it is the only freely available HMM-based predictor for beta-barrel outer membrane protein topology.
在全序列基因组中,整合膜蛋白约占所有蛋白质的20% - 30%。它们分为两种结构类型,即α - 螺旋膜蛋白和β - 桶状膜蛋白,具有不同的物理化学特性、结构和定位。虽然如今预测α - 螺旋整合膜蛋白的跨膜片段似乎是一项容易的任务,但对于β - 桶状膜蛋白来说则困难得多。我们基于隐马尔可夫模型开发了一种方法,能够预测革兰氏阴性菌外膜蛋白的跨膜β链,并在大型数据集中将其与水溶性蛋白区分开来。该模型以判别方式进行训练,旨在最大化正确预测的概率而非序列的似然性。
训练是在一个包含14个具有原子分辨率结构的外膜蛋白的非冗余数据库上进行的;通过留一法进行测试,每个残基的准确率为84.2%,相关系数为0.72,而在自一致性测试中,每个残基的准确率为88.1%,相关系数为0.824。在自一致性测试中,正确预测的拓扑结构总数为14个中的10个,留一法测试中为14个中的9个。此外,该模型能够在大规模应用中区分外膜蛋白和水溶性蛋白,外膜蛋白和水溶性蛋白正确分类的成功率分别为88.8%和89.2%,这是文献中报道的最高比率之一。该测试是在一组彼此之间以及与训练集蛋白质序列同一性较低的已知外膜蛋白上独立进行的。
基于上述结果,我们开发了一种策略,能够筛选大肠杆菌的整个蛋白质组以寻找外膜蛋白。结果令人满意,因此本文提出的方法似乎适用于筛选整个蛋白质组以发现新型外膜蛋白。非商业用户可通过以下网址访问网络界面:http://bioinformatics.biol.uoa.gr/PRED-TMBB,它是唯一免费的基于隐马尔可夫模型的β - 桶状外膜蛋白拓扑结构预测器。