Price Morgan N, Huang Katherine H, Alm Eric J, Arkin Adam P
Lawrence Berkeley National Lab 1 Cyclotron Road, Mailstop 939R704, Berkeley, CA 94720, USA.
Nucleic Acids Res. 2005 Feb 8;33(3):880-92. doi: 10.1093/nar/gki232. Print 2005.
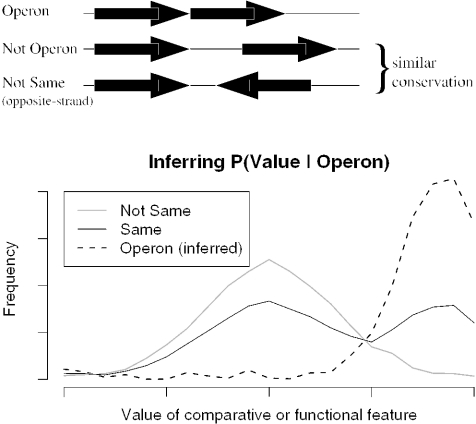
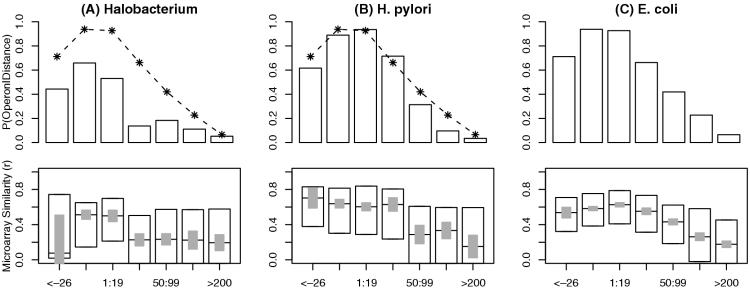
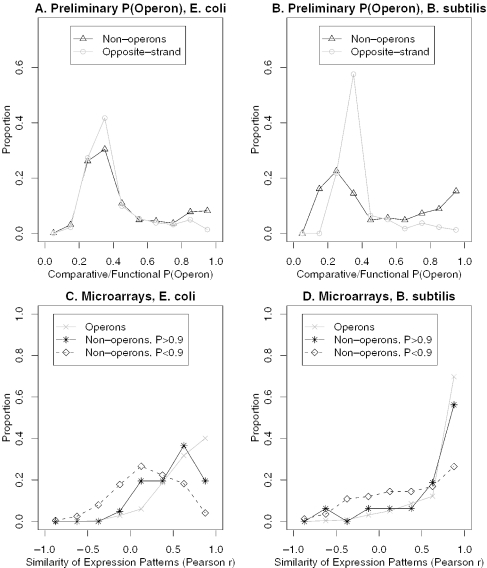
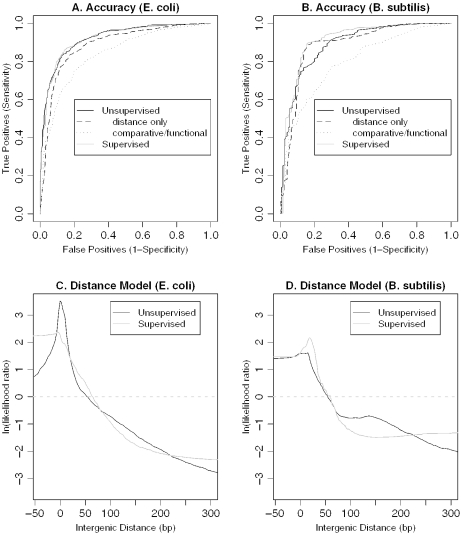
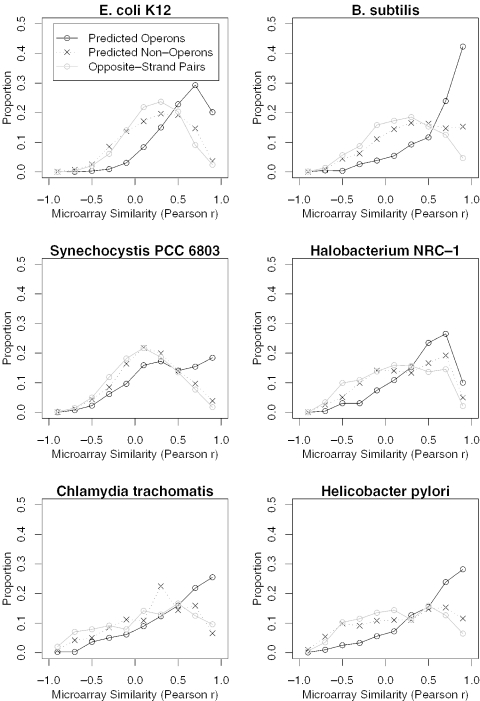
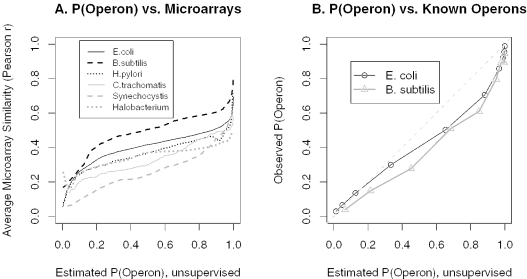
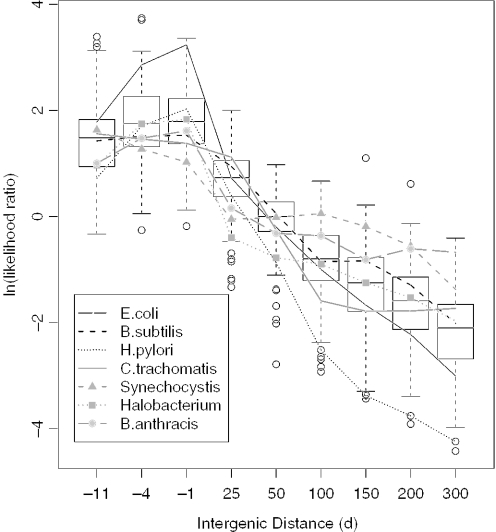
We combine comparative genomic measures and the distance separating adjacent genes to predict operons in 124 completely sequenced prokaryotic genomes. Our method automatically tailors itself to each genome using sequence information alone, and thus can be applied to any prokaryote. For Escherichia coli K12 and Bacillus subtilis, our method is 85 and 83% accurate, respectively, which is similar to the accuracy of methods that use the same features but are trained on experimentally characterized transcripts. In Halobacterium NRC-1 and in Helicobacter pylori, our method correctly infers that genes in operons are separated by shorter distances than they are in E.coli, and its predictions using distance alone are more accurate than distance-only predictions trained on a database of E.coli transcripts. We use microarray data from six phylogenetically diverse prokaryotes to show that combining intergenic distance with comparative genomic measures further improves accuracy and that our method is broadly effective. Finally, we survey operon structure across 124 genomes, and find several surprises: H.pylori has many operons, contrary to previous reports; Bacillus anthracis has an unusual number of pseudogenes within conserved operons; and Synechocystis PCC 6803 has many operons even though it has unusually wide spacings between conserved adjacent genes.
我们结合比较基因组学方法和相邻基因之间的间隔距离,来预测124个已完成全序列测定的原核生物基因组中的操纵子。我们的方法仅使用序列信息就能自动适应每个基因组,因此可应用于任何原核生物。对于大肠杆菌K12和枯草芽孢杆菌,我们的方法准确率分别为85%和83%,这与使用相同特征但基于实验确定的转录本进行训练的方法的准确率相似。在嗜盐菌NRC-1和幽门螺杆菌中,我们的方法正确推断出操纵子中的基因之间的间隔距离比在大肠杆菌中更短,并且仅使用距离进行的预测比基于大肠杆菌转录本数据库训练的仅距离预测更准确。我们使用来自六种系统发育上不同的原核生物的微阵列数据表明,将基因间距离与比较基因组学方法相结合可进一步提高准确率,并且我们的方法具有广泛的有效性。最后,我们调查了124个基因组的操纵子结构,发现了几个意外情况:与之前的报道相反,幽门螺杆菌有许多操纵子;炭疽芽孢杆菌在保守操纵子内有异常数量的假基因;集胞藻PCC 6803尽管在保守的相邻基因之间有异常宽的间隔,但仍有许多操纵子。