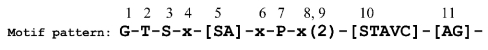Gutman Roee, Berezin Carine, Wollman Roy, Rosenberg Yossi, Ben-Tal Nir
Department of Biochemistry, The George S. Wise Faculty of Life Sciences, Tel Aviv University, Ramat Aviv 69978, Israel.
Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W255-61. doi: 10.1093/nar/gki496.
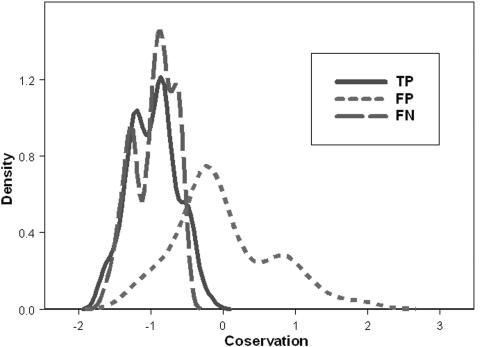
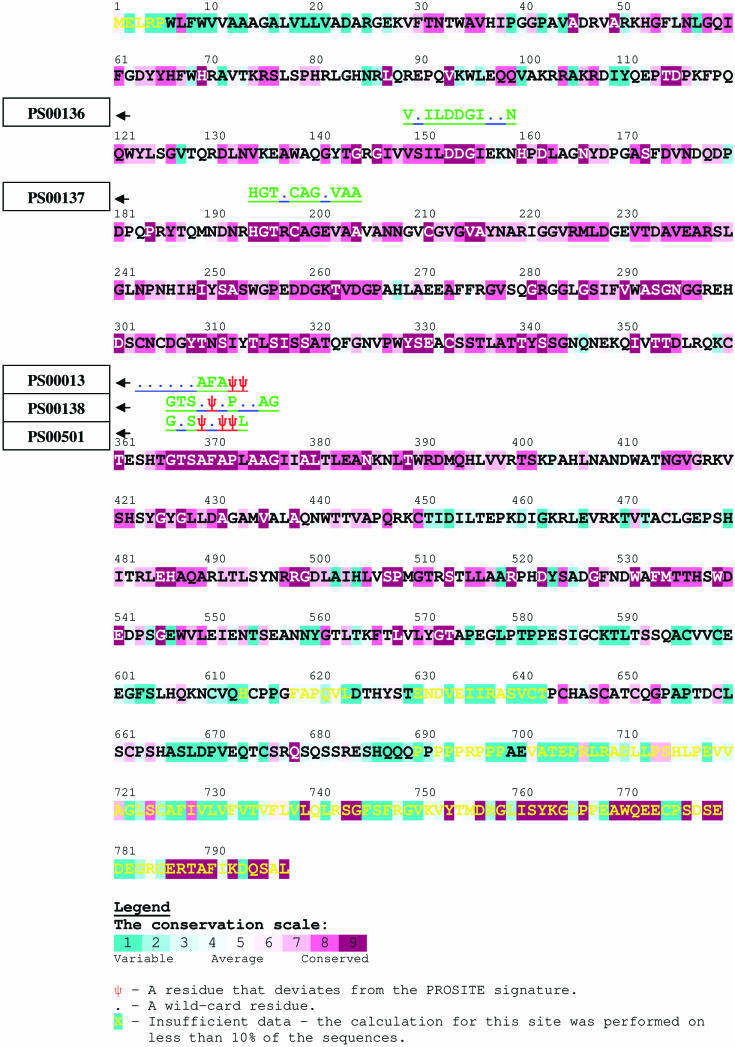
Sequence signature databases such as PROSITE, which include amino acid segments that are indicative of a protein's function, are useful for protein annotation. Lamentably, the annotation is not always accurate. A signature may be falsely detected in a protein that does not carry out the associated function (false positive prediction, FP) or may be overlooked in a protein that does carry out the function (false negative prediction, FN). A new approach has emerged in which a signature is replaced with a sequence profile, calculated based on multiple sequence alignment (MSA) of homologous proteins that share the same function. This approach, which is superior to the simple pattern search, essentially searches with the sequence of the query protein against an MSA library. We suggest here an alternative approach, implemented in the QuasiMotiFinder web server (http://quasimotifinder.tau.ac.il/), which is based on a search with an MSA of homologous query proteins against the original PROSITE signatures. The explicit use of the average evolutionary conservation of the signature in the query proteins significantly reduces the rate of FP prediction compared with the simple pattern search. QuasiMotiFinder also has a reduced rate of FN prediction compared with simple pattern searches, since the traditional search for precise signatures has been replaced by a permissive search for signature-like patterns that are physicochemically similar to known signatures. Overall, QuasiMotiFinder and the profile search are comparable to each other in terms of performance. They are also complementary to each other in that signatures that are falsely detected in (or overlooked by) one may be correctly detected by the other.
诸如PROSITE之类的序列特征数据库,其中包含指示蛋白质功能的氨基酸片段,对蛋白质注释很有用。遗憾的是,注释并不总是准确的。一个特征可能在不执行相关功能的蛋白质中被错误检测到(假阳性预测,FP),或者可能在执行该功能的蛋白质中被忽略(假阴性预测,FN)。一种新方法出现了,即用基于具有相同功能的同源蛋白质的多序列比对(MSA)计算的序列概况来取代特征。这种方法优于简单的模式搜索,本质上是用查询蛋白质的序列与一个MSA库进行比对。我们在此提出一种替代方法,在QuasiMotiFinder网络服务器(http://quasimotifinder.tau.ac.il/)中实现,该方法基于用同源查询蛋白质的MSA与原始PROSITE特征进行比对。与简单的模式搜索相比,明确使用查询蛋白质中特征的平均进化保守性可显著降低FP预测率。与简单模式搜索相比,QuasiMotiFinder的FN预测率也有所降低,因为传统的精确特征搜索已被对与已知特征在物理化学上相似的特征样模式的宽松搜索所取代。总体而言,QuasiMotiFinder和概况搜索在性能方面相互可比。它们也相互补充,因为在一个中被错误检测到(或被忽略)的特征可能会被另一个正确检测到。