Capriotti Emidio, Fariselli Piero, Casadio Rita
Laboratory of Biocomputing, CIRB/Department of Biology, University of Bologna via Irnerio 42, 40126 Bologna, Italy.
Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W306-10. doi: 10.1093/nar/gki375.
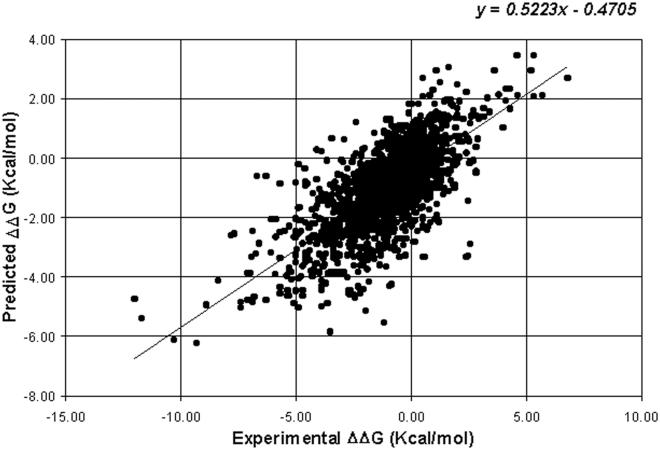
I-Mutant2.0 is a support vector machine (SVM)-based tool for the automatic prediction of protein stability changes upon single point mutations. I-Mutant2.0 predictions are performed starting either from the protein structure or, more importantly, from the protein sequence. This latter task, to the best of our knowledge, is exploited for the first time. The method was trained and tested on a data set derived from ProTherm, which is presently the most comprehensive available database of thermodynamic experimental data of free energy changes of protein stability upon mutation under different conditions. I-Mutant2.0 can be used both as a classifier for predicting the sign of the protein stability change upon mutation and as a regression estimator for predicting the related DeltaDeltaG values. Acting as a classifier, I-Mutant2.0 correctly predicts (with a cross-validation procedure) 80% or 77% of the data set, depending on the usage of structural or sequence information, respectively. When predicting DeltaDeltaG values associated with mutations, the correlation of predicted with expected/experimental values is 0.71 (with a standard error of 1.30 kcal/mol) and 0.62 (with a standard error of 1.45 kcal/mol) when structural or sequence information are respectively adopted. Our web interface allows the selection of a predictive mode that depends on the availability of the protein structure and/or sequence. In this latter case, the web server requires only pasting of a protein sequence in a raw format. We therefore introduce I-Mutant2.0 as a unique and valuable helper for protein design, even when the protein structure is not yet known with atomic resolution.
http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi.
I-Mutant2.0是一种基于支持向量机(SVM)的工具,用于自动预测单点突变后蛋白质稳定性的变化。I-Mutant2.0的预测可以从蛋白质结构开始,或者更重要的是,从蛋白质序列开始。据我们所知,后一项任务是首次被利用。该方法在一个源自ProTherm的数据集上进行了训练和测试,ProTherm是目前关于不同条件下突变后蛋白质稳定性自由能变化的热力学实验数据最全面的可用数据库。I-Mutant2.0既可以用作预测突变后蛋白质稳定性变化符号的分类器,也可以用作预测相关ΔΔG值的回归估计器。作为分类器,I-Mutant2.0(通过交叉验证程序)分别根据结构或序列信息的使用情况,正确预测了数据集中80%或77%的数据。在预测与突变相关的ΔΔG值时,当分别采用结构或序列信息时,预测值与预期/实验值的相关性分别为0.71(标准误差为1.30千卡/摩尔)和0.62(标准误差为1.45千卡/摩尔)。我们的网络界面允许根据蛋白质结构和/或序列的可用性选择预测模式。在后一种情况下,网络服务器只需要粘贴原始格式的蛋白质序列。因此,即使蛋白质结构尚未达到原子分辨率,我们也将I-Mutant2.0作为蛋白质设计中一个独特且有价值的辅助工具引入。
http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi 。