Schymkowitz Joost, Borg Jesper, Stricher Francois, Nys Robby, Rousseau Frederic, Serrano Luis
Switch Laboratory, Flanders Interuniversity Institute for Biotechnology (VIB), Vrije Universiteit Brussel, Pleinlaan 2, 1050 Brussel, Belgium.
Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W382-8. doi: 10.1093/nar/gki387.
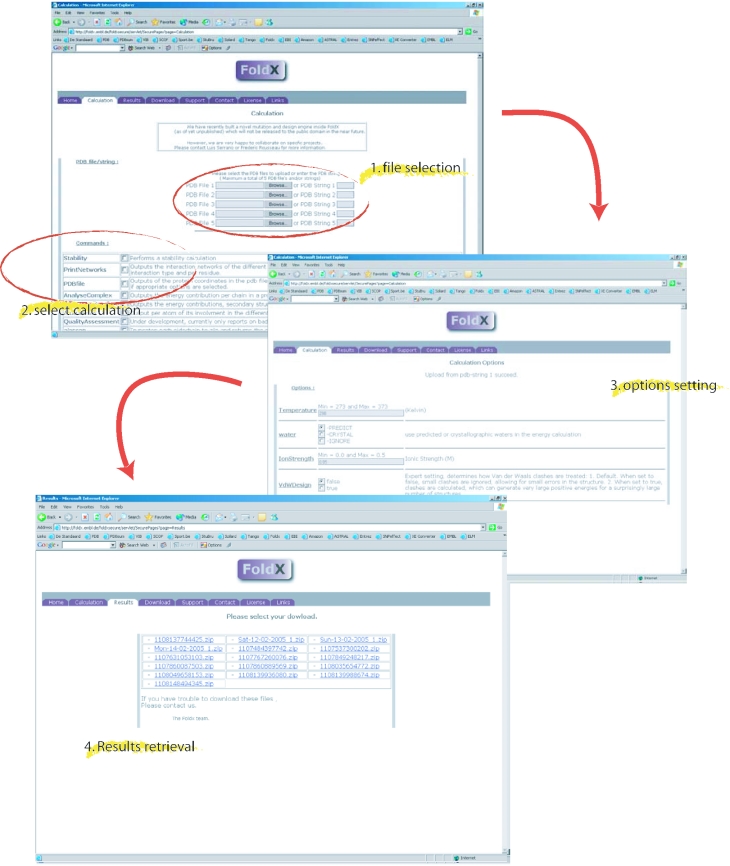
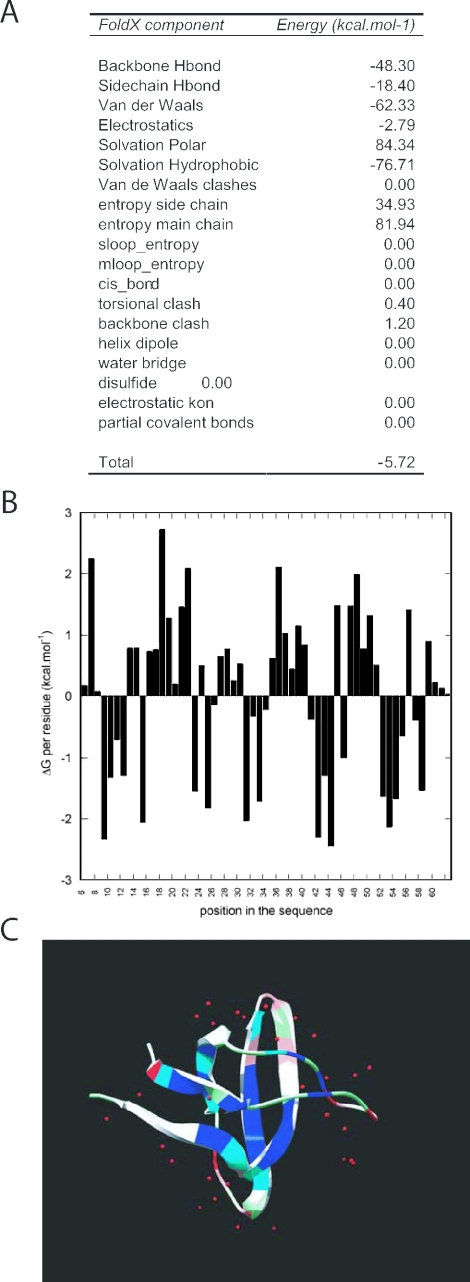
FoldX is an empirical force field that was developed for the rapid evaluation of the effect of mutations on the stability, folding and dynamics of proteins and nucleic acids. The core functionality of FoldX, namely the calculation of the free energy of a macromolecule based on its high-resolution 3D structure, is now publicly available through a web server at http://foldx.embl.de/. The current release allows the calculation of the stability of a protein, calculation of the positions of the protons and the prediction of water bridges, prediction of metal binding sites and the analysis of the free energy of complex formation. Alanine scanning, the systematic truncation of side chains to alanine, is also included. In addition, some reporting functions have been added, and it is now possible to print both the atomic interaction networks that constitute the protein, print the structural and energetic details of the interactions per atom or per residue, as well as generate a general quality report of the pdb structure. This core functionality will be further extended as more FoldX applications are developed.
FoldX是一种经验性力场,旨在快速评估突变对蛋白质和核酸的稳定性、折叠及动力学的影响。FoldX的核心功能,即基于大分子的高分辨率三维结构计算其自由能,现在可通过http://foldx.embl.de/的网络服务器公开获取。当前版本允许计算蛋白质的稳定性、质子位置预测及水桥预测、金属结合位点预测以及复合物形成自由能分析。还包括丙氨酸扫描,即将侧链系统截短为丙氨酸。此外,添加了一些报告功能,现在可以打印构成蛋白质的原子相互作用网络、打印每个原子或每个残基相互作用的结构和能量细节,以及生成pdb结构的总体质量报告。随着更多FoldX应用的开发,这一核心功能将得到进一步扩展。