Jacobson Kenneth A, Costanzi Stefano, Ivanov Andrei A, Tchilibon Susanna, Besada Pedro, Gao Zhan-Guo, Maddileti Savitri, Harden T Kendall
Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Biochem Pharmacol. 2006 Feb 14;71(4):540-9. doi: 10.1016/j.bcp.2005.11.010. Epub 2005 Dec 15.
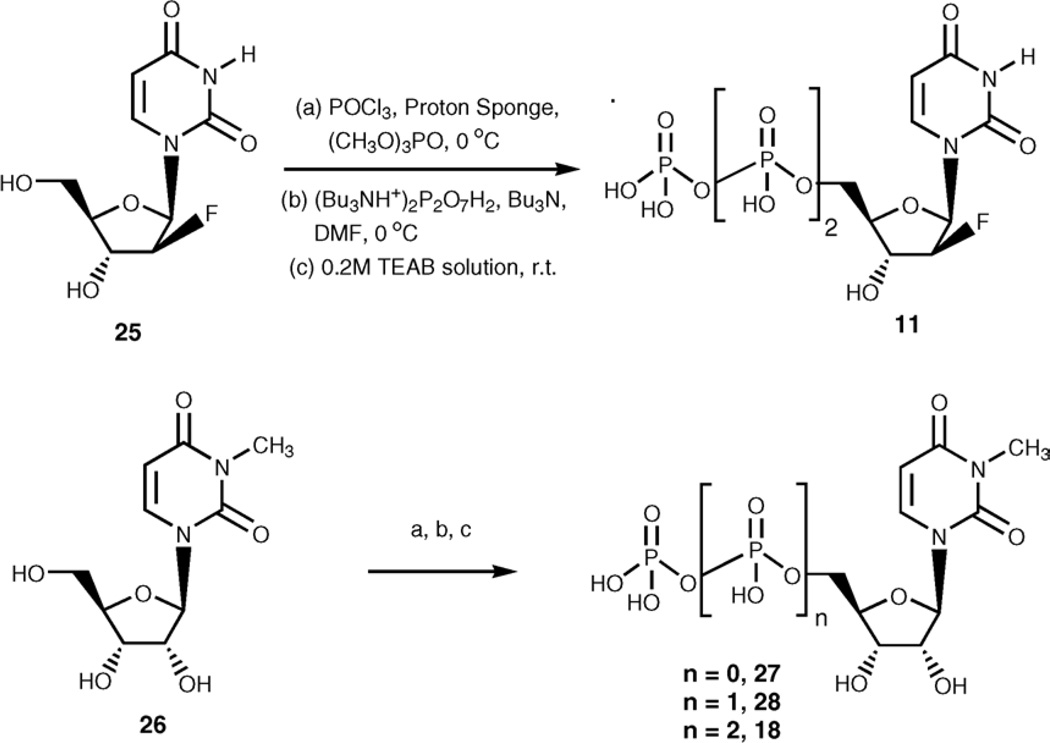
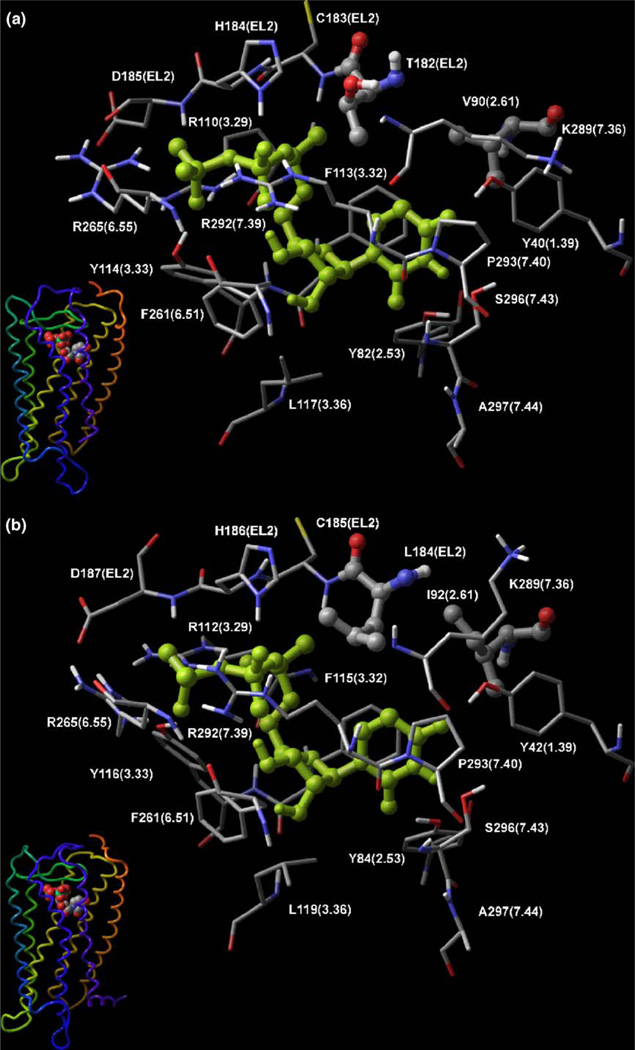
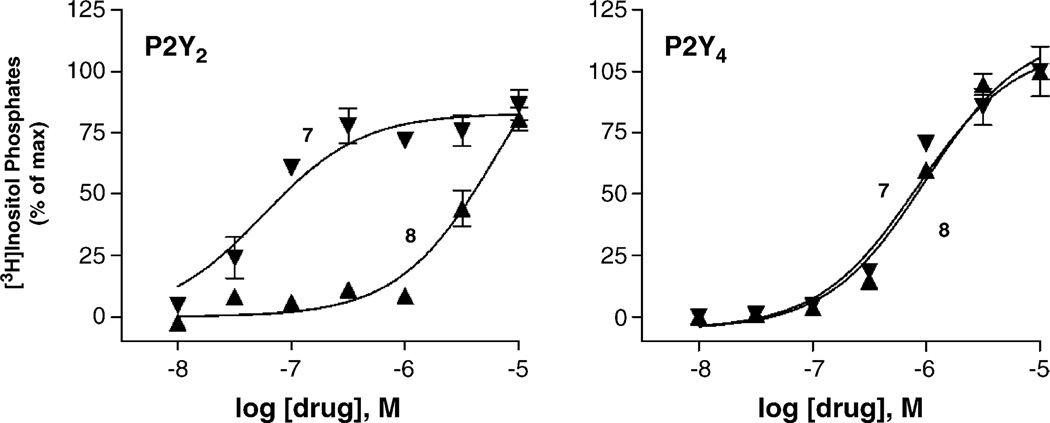
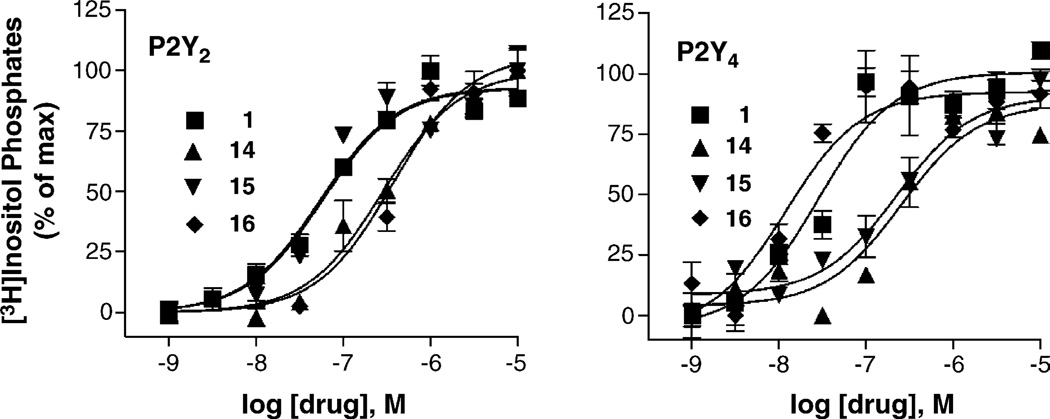
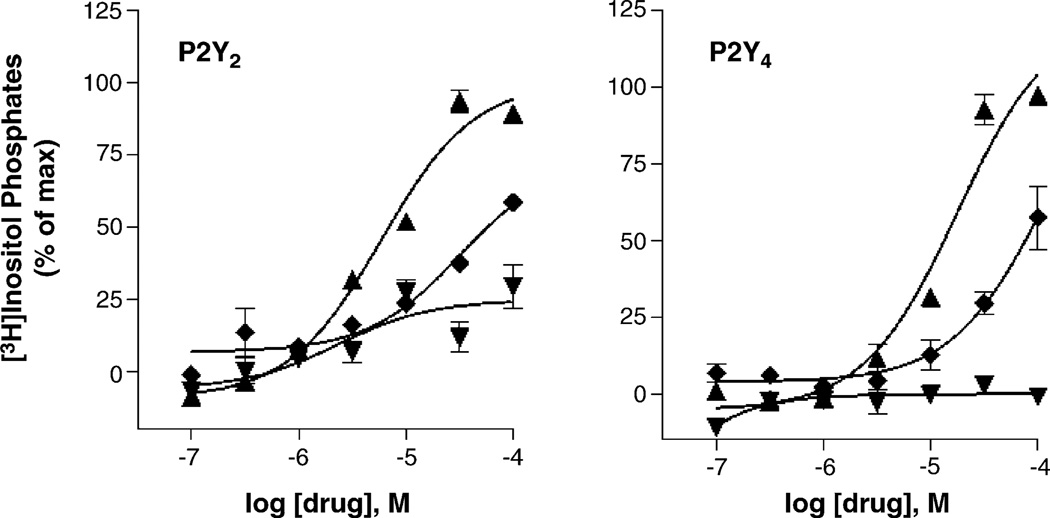
With the long-term goal of developing receptor subtype-selective high affinity agonists for the uracil nucleotide-activated P2Y receptors we have carried out a series of structure activity and molecular modeling studies of the human P2Y2 and P2Y4 receptors. UTP analogues with substitutions in the 2'-position of the ribose moiety retained capacity to activate both P2Y2 and P2Y4 receptors. Certain of these analogues were equieffective for activation of both receptors whereas 2'-amino-2'-deoxy-UTP exhibited higher potency for the P2Y2 receptor and 2'-azido-UTP exhibited higher potency for the P2Y4 receptor. 4-Thio substitution of the uracil base resulted in a UTP analogue with increased potency relative to UTP for activation of both the P2Y2 and P2Y4 receptors. In contrast, 2-thio substitution and halo- or alkyl substitution in the 5-position of the uracil base resulted in molecules that were 3-30-fold more potent at the P2Y2 receptor than P2Y4 receptor. 6-Aza-UTP was a P2Y2 receptor agonist that exhibited no activity at the P2Y4 receptor. Stereoisomers of UTPalphaS and 2'-deoxy-UTPalphaS were more potent at the P2Y2 than P2Y4 receptor, and the R-configuration was favored at both receptors. Molecular docking studies revealed that the binding mode of UTP is similar for both the P2Y2 and P2Y4 receptor binding pockets with the most prominent dissimilarities of the two receptors located in the second transmembrane domain (V90 in the P2Y2 receptor and I92 in the P2Y4 receptor) and the second extracellular loop (T182 in the P2Y2 receptor and L184 in the P2Y4 receptor). In summary, this work reveals substitutions in UTP that differentially affect agonist activity at P2Y2 versus P2Y4 receptors and in combination with molecular modeling studies should lead to chemical synthesis of new receptor subtype-selective drugs.
为了实现开发尿嘧啶核苷酸激活的P2Y受体亚型选择性高亲和力激动剂的长期目标,我们对人P2Y2和P2Y4受体进行了一系列构效关系和分子模拟研究。核糖部分2'-位有取代的UTP类似物保留了激活P2Y2和P2Y4受体的能力。其中某些类似物对两种受体的激活效果相同,而2'-氨基-2'-脱氧-UTP对P2Y2受体表现出更高的效力,2'-叠氮基-UTP对P2Y4受体表现出更高的效力。尿嘧啶碱基的4-硫代取代产生了一种相对于UTP对P2Y2和P2Y4受体激活效力均增加的UTP类似物。相比之下,尿嘧啶碱基5-位的2-硫代取代以及卤代或烷基取代产生的分子对P2Y2受体的效力比对P2Y4受体高3至30倍。6-氮杂-UTP是一种P2Y2受体激动剂,对P2Y4受体无活性。UTPαS和2'-脱氧-UTPαS的立体异构体对P2Y2受体的效力比对P2Y4受体更强,并且两种受体均更倾向于R构型。分子对接研究表明,UTP在P2Y2和P2Y4受体结合口袋中的结合模式相似,两种受体最显著的差异位于第二个跨膜结构域(P2Y2受体中的V90和P2Y4受体中的I92)和第二个细胞外环(P2Y2受体中的T182和P2Y4受体中的L184)。总之,这项工作揭示了UTP中的取代对P2Y2和P2Y4受体激动剂活性有不同影响,结合分子模拟研究应能导向新型受体亚型选择性药物的化学合成。