Ting Jason C, Ye Ying, Thomas George H, Ruczinski Ingo, Pevsner Jonathan
Department of Neurology, Kennedy Krieger Institute, Baltimore, Maryland 21205, USA.
BMC Bioinformatics. 2006 Jan 18;7:25. doi: 10.1186/1471-2105-7-25.
A variety of diseases are caused by chromosomal abnormalities such as aneuploidies (having an abnormal number of chromosomes), microdeletions, microduplications, and uniparental disomy. High density single nucleotide polymorphism (SNP) microarrays provide information on chromosomal copy number changes, as well as genotype (heterozygosity and homozygosity). SNP array studies generate multiple types of data for each SNP site, some with more than 100,000 SNPs represented on each array. The identification of different classes of anomalies within SNP data has been challenging.
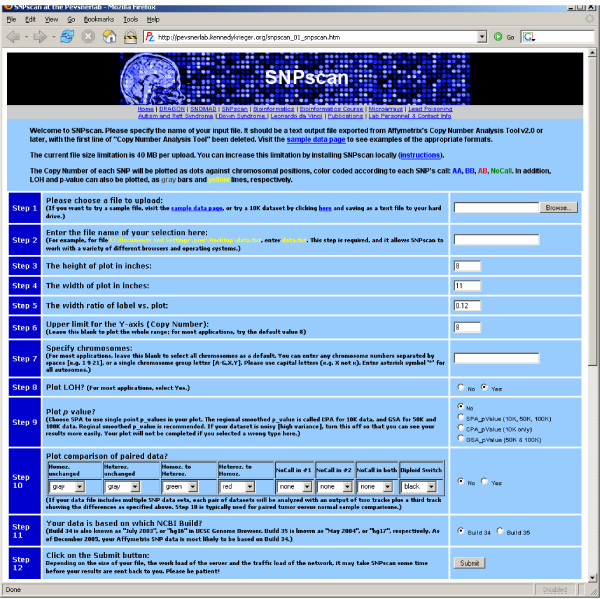
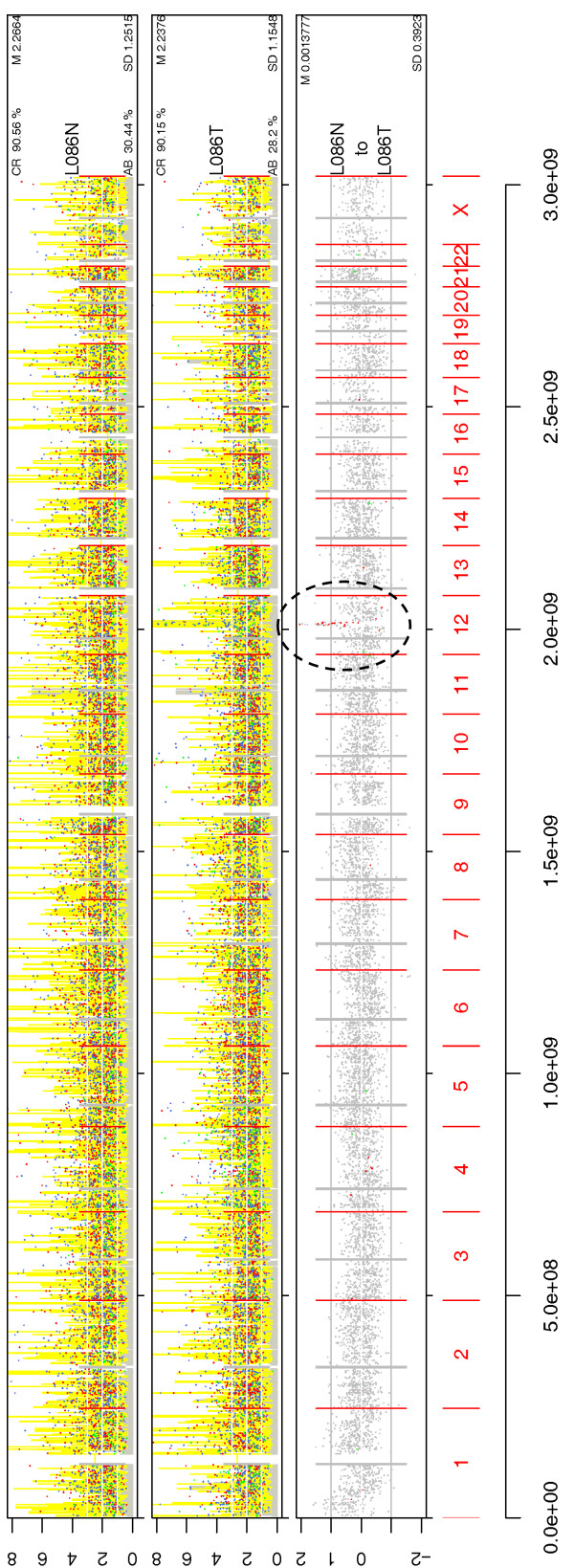
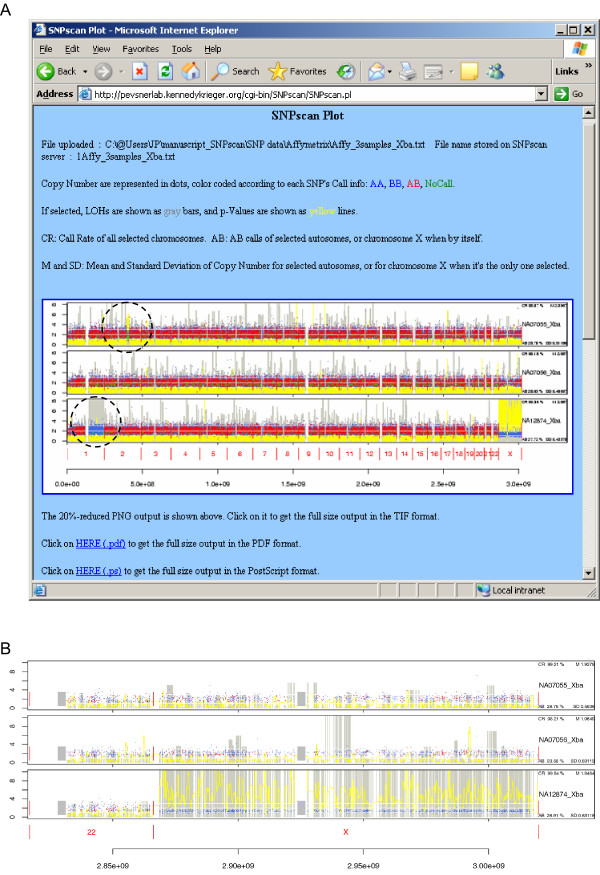
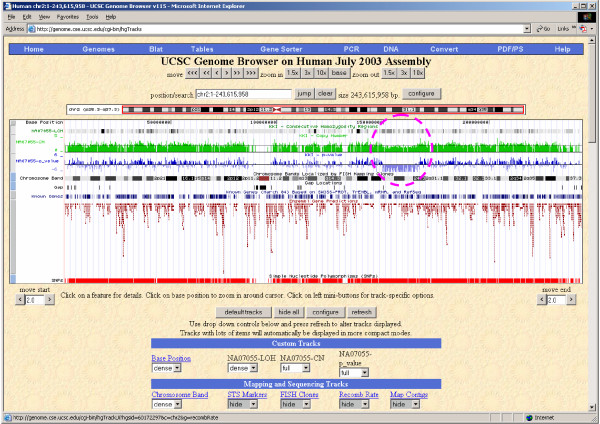
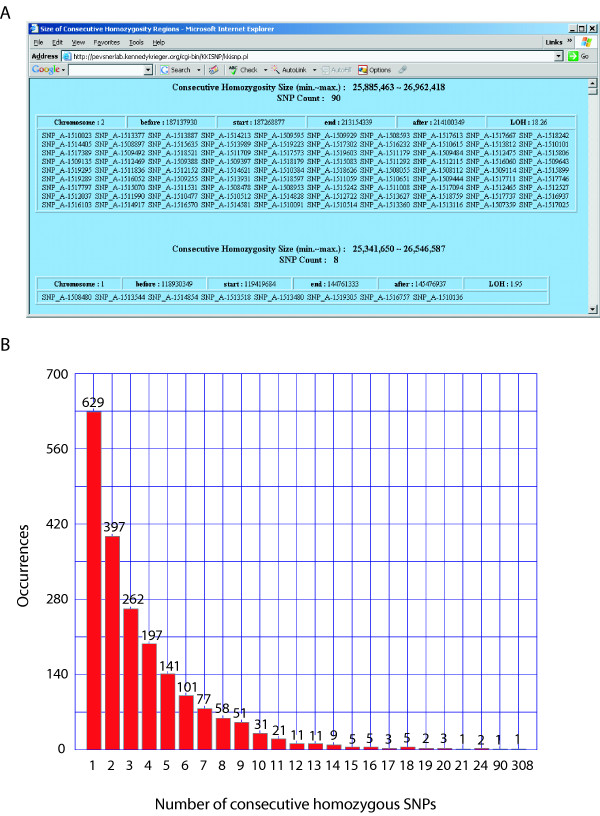
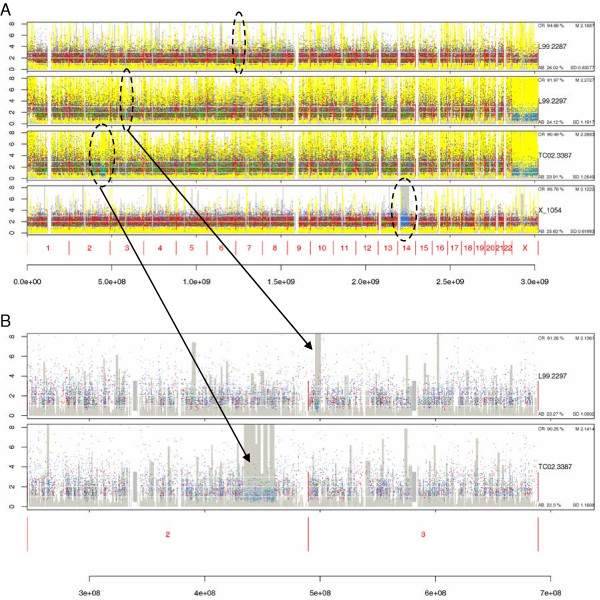
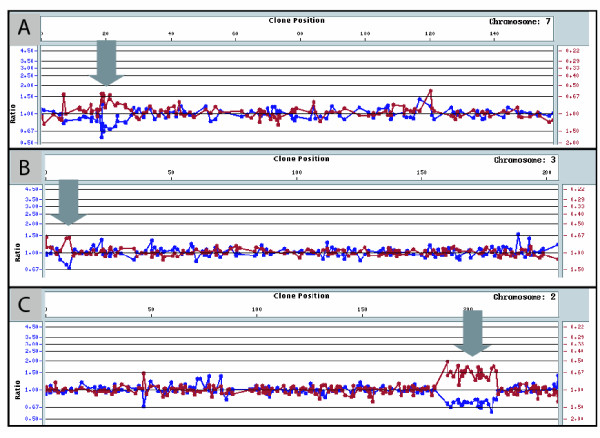
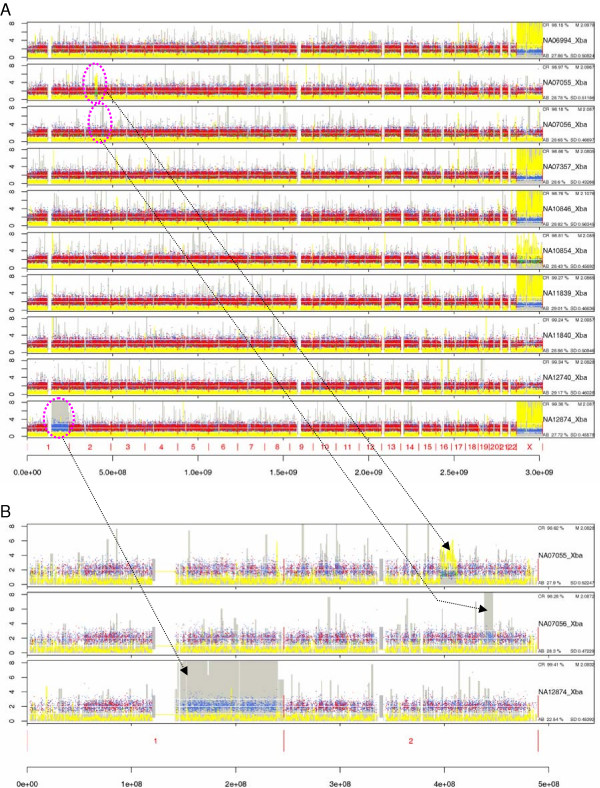
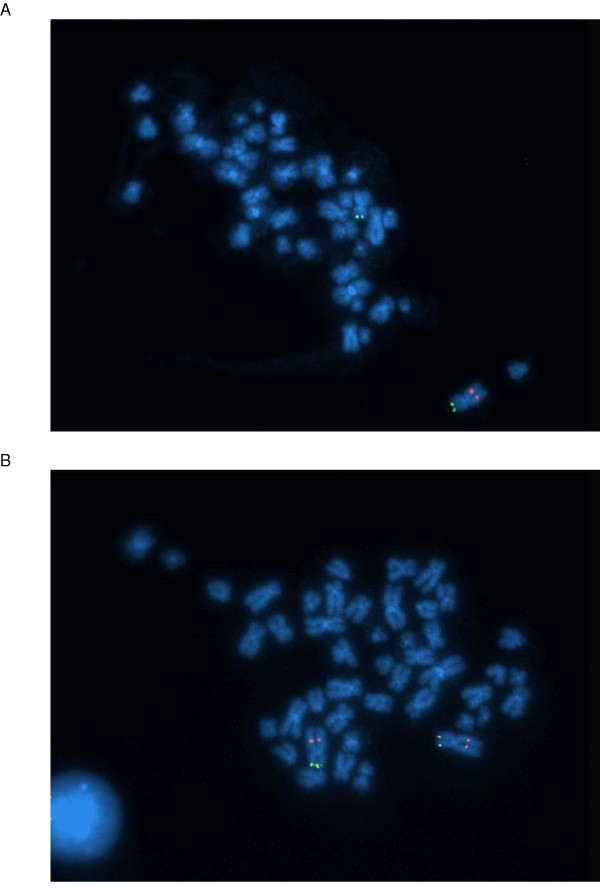
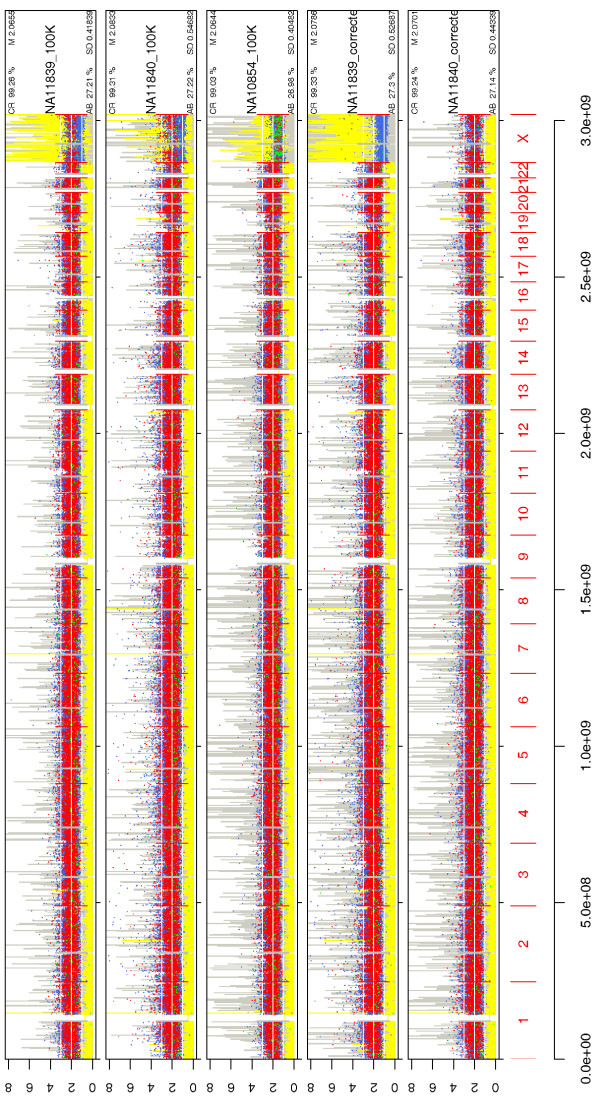
We have developed SNPscan, a web-accessible tool to analyze and visualize high density SNP data. It enables researchers (1) to visually and quantitatively assess the quality of user-generated SNP data relative to a benchmark data set derived from a control population, (2) to display SNP intensity and allelic call data in order to detect chromosomal copy number anomalies (duplications and deletions), (3) to display uniparental isodisomy based on loss of heterozygosity (LOH) across genomic regions, (4) to compare paired samples (e.g. tumor and normal), and (5) to generate a file type for viewing SNP data in the University of California, Santa Cruz (UCSC) Human Genome Browser. SNPscan accepts data exported from Affymetrix Copy Number Analysis Tool as its input. We validated SNPscan using data generated from patients with known deletions, duplications, and uniparental disomy. We also inspected previously generated SNP data from 90 apparently normal individuals from the Centre d'Etude du Polymorphisme Humain (CEPH) collection, and identified three cases of uniparental isodisomy, four females having an apparently mosaic X chromosome, two mislabelled SNP data sets, and one microdeletion on chromosome 2 with mosaicism from an apparently normal female. These previously unrecognized abnormalities were all detected using SNPscan. The microdeletion was independently confirmed by fluorescence in situ hybridization, and a region of homozygosity in a UPD case was confirmed by sequencing of genomic DNA.
SNPscan is useful to identify chromosomal abnormalities based on SNP intensity (such as chromosomal copy number changes) and heterozygosity data (including regions of LOH and some cases of UPD). The program and source code are available at the SNPscan website http://pevsnerlab.kennedykrieger.org/snpscan.htm.
多种疾病由染色体异常引起,如非整倍体(染色体数目异常)、微缺失、微重复和单亲二体。高密度单核苷酸多态性(SNP)微阵列可提供有关染色体拷贝数变化以及基因型(杂合性和纯合性)的信息。SNP阵列研究为每个SNP位点生成多种类型的数据,每个阵列上有超过100,000个SNP。在SNP数据中识别不同类型的异常具有挑战性。
我们开发了SNPscan,这是一个可通过网络访问的工具,用于分析和可视化高密度SNP数据。它使研究人员能够:(1)相对于来自对照人群的基准数据集,直观且定量地评估用户生成的SNP数据的质量;(2)显示SNP强度和等位基因分型数据,以检测染色体拷贝数异常(重复和缺失);(3)基于基因组区域的杂合性缺失(LOH)显示单亲同二体;(4)比较配对样本(如肿瘤和正常样本);(5)生成一种文件类型,以便在加利福尼亚大学圣克鲁兹分校(UCSC)人类基因组浏览器中查看SNP数据。SNPscan接受从Affymetrix拷贝数分析工具导出的数据作为输入。我们使用来自已知缺失、重复和单亲二体患者的数据验证了SNPscan。我们还检查了先前从人类多态性研究中心(CEPH)收集的90名明显正常个体生成的SNP数据,识别出3例单亲同二体、4名具有明显嵌合X染色体的女性、2个标记错误的SNP数据集,以及1例来自一名明显正常女性的2号染色体微缺失且伴有嵌合现象。这些先前未被识别的异常均通过SNPscan检测到。微缺失通过荧光原位杂交独立确认,单亲二体病例中的纯合区域通过基因组DNA测序确认。
SNPscan有助于基于SNP强度(如染色体拷贝数变化)和杂合性数据(包括LOH区域和一些单亲二体病例)识别染色体异常。该程序和源代码可在SNPscan网站http://pevsnerlab.kennedykrieger.org/snpscan.htm获取。