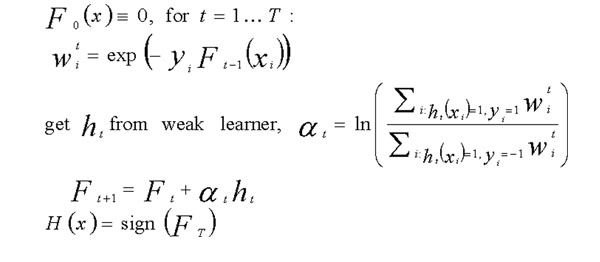HCNR Center for Bioinformatics, Brigham and Women's Hospital, Harvard Medical School, Boston, MA 02215, USA.
BMC Genet. 2005 Dec 30;6 Suppl 1(Suppl 1):S132. doi: 10.1186/1471-2156-6-S1-S132.
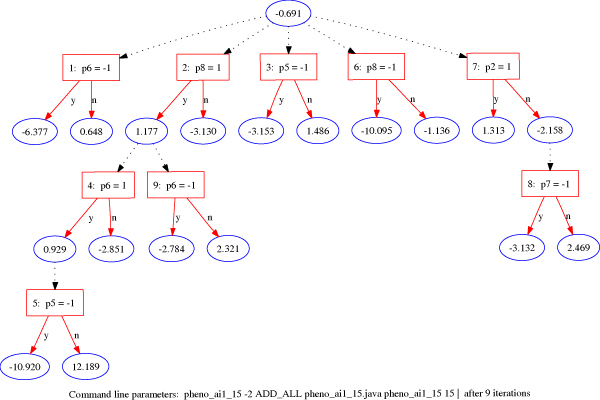
We applied the alternating decision trees (ADTrees) method to the last 3 replicates from the Aipotu, Danacca, Karangar, and NYC populations in the Problem 2 simulated Genetic Analysis Workshop dataset. Using information from the 12 binary phenotypes and sex as input and Kofendrerd Personality Disorder disease status as the outcome of ADTrees-based classifiers, we obtained a new quantitative trait based on average prediction scores, which was then used for genome-wide quantitative trait linkage (QTL) analysis. ADTrees are machine learning methods that combine boosting and decision trees algorithms to generate smaller and easier-to-interpret classification rules. In this application, we compared four modeling strategies from the combinations of two boosting iterations (log or exponential loss functions) coupled with two choices of tree generation types (a full alternating decision tree or a classic boosting decision tree). These four different strategies were applied to the founders in each population to construct four classifiers, which were then applied to each study participant. To compute average prediction score for each subject with a specific trait profile, such a process was repeated with 10 runs of 10-fold cross validation, and standardized prediction scores obtained from the 10 runs were averaged and used in subsequent expectation-maximization Haseman-Elston QTL analyses (implemented in GENEHUNTER) with the approximate 900 SNPs in Hardy-Weinberg equilibrium provided for each population. Our QTL analyses on the basis of four models (a full alternating decision tree and a classic boosting decision tree paired with either log or exponential loss function) detected evidence for linkage (Z >or= 1.96, p < 0.01) on chromosomes 1, 3, 5, and 9. Moreover, using average iteration and abundance scores for the 12 phenotypes and sex as their relevancy measurements, we found all relevant phenotypes for all four populations except phenotype b for the Karangar population, with suggested subgroup structure consistent with latent traits used in the model. In conclusion, our findings suggest that the ADTrees method may offer a more accurate representation of the disease status that allows for better detection of linkage evidence.
我们将交替决策树(ADTrees)方法应用于问题 2 模拟遗传分析研讨会数据集中 Aipotu、Danacca、Karangar 和 NYC 群体的最后 3 个重复。使用 12 个二元表型和性别信息作为输入,以 Kofendrerd 人格障碍疾病状态作为 ADTrees 分类器的输出,我们获得了一个新的基于平均预测分数的数量性状,然后用于全基因组数量性状连锁(QTL)分析。ADTrees 是一种机器学习方法,它结合了提升和决策树算法,生成更小、更易于解释的分类规则。在这种应用中,我们比较了四种来自两种提升迭代(对数或指数损失函数)与两种树生成类型(完全交替决策树或经典提升决策树)组合的建模策略。这四种不同的策略应用于每个群体的创始人,构建了四个分类器,然后将这些分类器应用于每个研究参与者。为了计算具有特定特征谱的每个个体的平均预测分数,我们通过 10 次 10 折交叉验证重复该过程,从 10 次运行中获得的标准化预测分数平均,并用于后续期望最大化 Haseman-Elston QTL 分析(在 GENEHUNTER 中实现),对于每个群体,提供了大约 900 个处于哈迪-温伯格平衡的 SNP。我们基于四种模型(完全交替决策树和经典提升决策树与对数或指数损失函数配对)的 QTL 分析检测到染色体 1、3、5 和 9 上存在连锁证据(Z≥1.96,p<0.01)。此外,使用 12 个表型和性别的平均迭代和丰度分数作为它们的相关性测量,我们发现了除了 Karangar 群体的表型 b 之外,所有四个群体的所有相关表型,具有与模型中使用的潜在特征一致的建议亚群结构。总之,我们的研究结果表明,ADTrees 方法可能提供了对疾病状态更准确的表示,从而更好地检测连锁证据。