Department of Epidemiology and Biostatistics, Wolstein Research Building, Case Western Reserve University, Cleveland, OH 44106, USA.
BMC Genet. 2005 Dec 30;6 Suppl 1(Suppl 1):S29. doi: 10.1186/1471-2156-6-S1-S29.
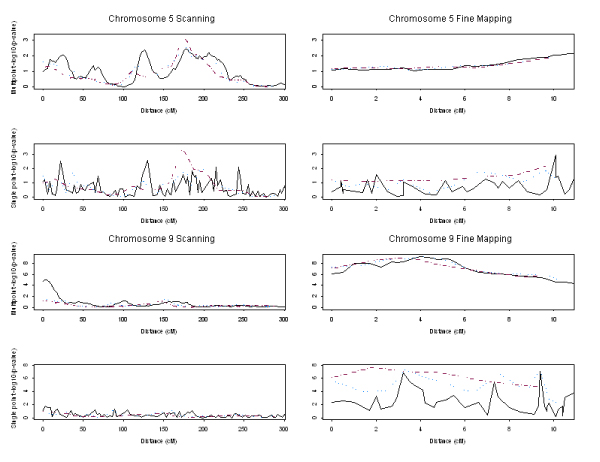
There is growing evidence that a map of dense single-nucleotide polymorphisms (SNPs) can outperform a map of sparse microsatellites for linkage analysis. There is also argument as to whether a clustered SNP map can outperform an evenly spaced SNP map. Using Genetic Analysis Workshop 14 simulated data, we compared for linkage analysis microsatellites, SNPs, and composite markers derived from SNPs. We encoded the composite markers in a two-step approach, in which the maximum identity length contrast method was employed to allow for recombination between loci. A SNP map 2.3 times as dense as a microsatellite map (approximately 2.9 cM compared to approximately 6.7 cM apart) provided slightly less information content (approximately 0.83 compared to approximately 0.89). Most inheritance information could be extracted when the SNPs were spaced < 1 cM apart. Comparing the linkage results on using SNPs or composite markers derived from them based on both 3 cM and 0.3 cM resolution maps, we showed that the inter-SNP distance should be kept small (< 1 cM), and that for multipoint linkage analysis the original markers and the derived composite markers had similar power; but for single point linkage analysis the resulting composite markers lead to more power. Considering all factors, such as information content, flexibility of analysis method, map errors, and genotyping errors, a map of clustered SNPs can be an efficient design for a genome-wide linkage scan.
越来越多的证据表明,密集的单核苷酸多态性 (SNP) 图谱在连锁分析中可以胜过稀疏的微卫星图谱。也有争议认为,聚类 SNP 图谱是否可以胜过均匀间隔的 SNP 图谱。我们使用遗传分析研讨会 14 模拟数据,比较了连锁分析中的微卫星、SNP 和源自 SNP 的复合标记。我们采用两步法对复合标记进行编码,其中最大同源性长度对比方法允许在基因座之间发生重组。SNP 图谱的密度是微卫星图谱的 2.3 倍(大约 2.9 cM 而不是大约 6.7 cM),提供的信息量略少(大约 0.83 而不是大约 0.89)。当 SNP 之间的间隔<1 cM 时,可以提取大部分遗传信息。根据 3 cM 和 0.3 cM 分辨率图谱,比较使用 SNP 或源自它们的复合标记进行连锁分析的结果,我们表明 SNP 之间的距离应该保持较小(<1 cM),对于多点连锁分析,原始标记和衍生的复合标记具有相似的效力;但是对于单点连锁分析,所得的复合标记会导致更高的效力。综合考虑所有因素,如信息量、分析方法的灵活性、图谱错误和基因分型错误,聚类 SNP 图谱可以成为全基因组连锁扫描的有效设计。