Mathe Ewy, Olivier Magali, Kato Shunsuke, Ishioka Chikashi, Hainaut Pierre, Tavtigian Sean V
International Agency for Research on Cancer, Lyon, France.
Nucleic Acids Res. 2006 Mar 6;34(5):1317-25. doi: 10.1093/nar/gkj518. Print 2006.
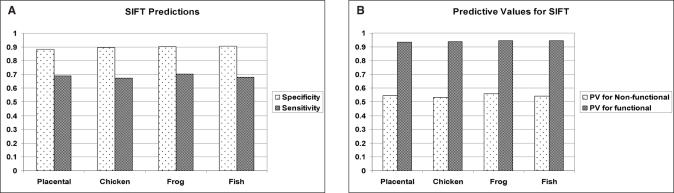
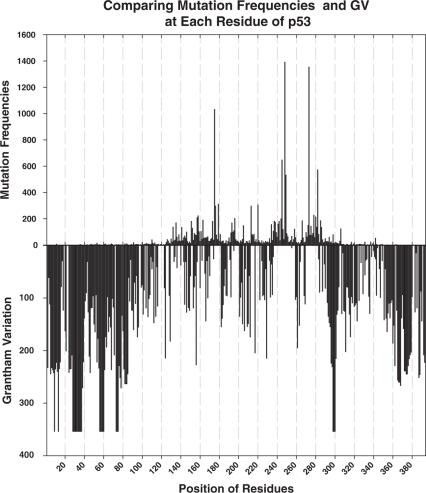
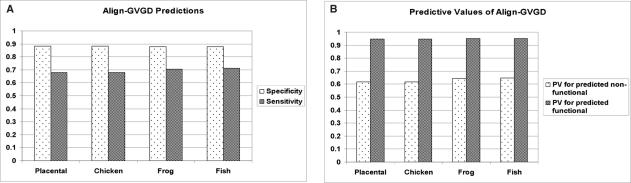
Prediction of the biological effect of missense substitutions has become important because they are often observed in known or candidate disease susceptibility genes. In this paper, we carried out a 3-step analysis of 1514 missense substitutions in the DNA-binding domain (DBD) of TP53, the most frequently mutated gene in human cancers. First, we calculated two types of conservation scores based on a TP53 multiple sequence alignment (MSA) for each substitution: (i) Grantham Variation (GV), which measures the degree of biochemical variation among amino acids found at a given position in the MSA; (ii) Grantham Deviation (GD), which reflects the 'biochemical distance' of the mutant amino acid from the observed amino acid at a particular position (given by GV). Second, we used a method that combines GV and GD scores, Align-GVGD, to predict the transactivation activity of each missense substitution. We compared our predictions against experimentally measured transactivation activity (yeast assays) to evaluate their accuracy. Finally, the prediction results were compared with those obtained by the program Sorting Intolerant from Tolerant (SIFT) and Dayhoff's classification. Our predictions yielded high prediction accuracy for mutants showing a loss of transactivation ( approximately 88% specificity) with lower prediction accuracy for mutants with transactivation similar to that of the wild-type (67.9 to 71.2% sensitivity). Align-GVGD results were comparable to SIFT (88.3 to 90.6% and 67.4 to 70.3% specificity and sensitivity, respectively) and outperformed Dayhoff's classification (80 and 40.9% specificity and sensitivity, respectively). These results further demonstrate the utility of the Align-GVGD method, which was previously applied to BRCA1. Align-GVGD is available online at http://agvgd.iarc.fr.
错义替换的生物学效应预测变得愈发重要,因为它们常在已知或候选疾病易感基因中被观察到。在本文中,我们对人类癌症中最常发生突变的基因TP53的DNA结合结构域(DBD)中的1514个错义替换进行了三步分析。首先,我们基于TP53多序列比对(MSA)为每个替换计算了两种类型的保守性得分:(i)格兰瑟姆变异(GV),它衡量了MSA中给定位置发现的氨基酸之间的生化变异程度;(ii)格兰瑟姆偏差(GD),它反映了突变氨基酸与特定位置观察到的氨基酸之间的“生化距离”(由GV给出)。其次,我们使用一种结合GV和GD得分的方法Align-GVGD来预测每个错义替换的反式激活活性。我们将我们的预测与实验测量的反式激活活性(酵母试验)进行比较,以评估其准确性。最后,将预测结果与通过容忍中不耐受排序程序(SIFT)和戴霍夫分类法获得的结果进行比较。对于显示反式激活丧失的突变体,我们的预测具有较高的预测准确性(特异性约为88%),而对于反式激活与野生型相似的突变体,预测准确性较低(敏感性为67.9%至71.2%)。Align-GVGD的结果与SIFT相当(特异性和敏感性分别为88.3%至90.6%和67.4%至70.3%),并且优于戴霍夫分类法(特异性和敏感性分别为80%和40.9%)。这些结果进一步证明了Align-GVGD方法的实用性,该方法先前已应用于BRCA1。Align-GVGD可在http://agvgd.iarc.fr在线获取。