Sadreyev Ruslan I, Grishin Nick V
Howard Hughes Medical Institute, Department of Biochemistry, University of Texas Southwestern Medical Center, Dallas, TX 75390-8816, USA.
BMC Struct Biol. 2006 Mar 20;6:6. doi: 10.1186/1472-6807-6-6.
As tertiary structure is currently available only for a fraction of known protein families, it is important to assess what parts of sequence space have been structurally characterized. We consider protein domains whose structure can be predicted by sequence similarity to proteins with solved structure and address the following questions. Do these domains represent an unbiased random sample of all sequence families? Do targets solved by structural genomic initiatives (SGI) provide such a sample? What are approximate total numbers of structure-based superfamilies and folds among soluble globular domains?
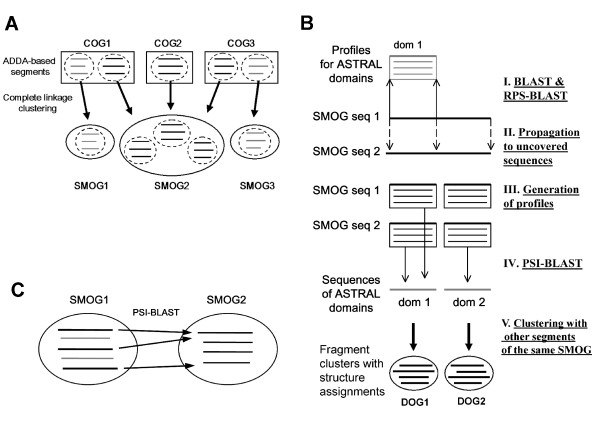
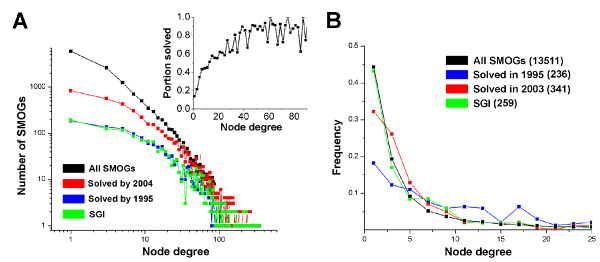
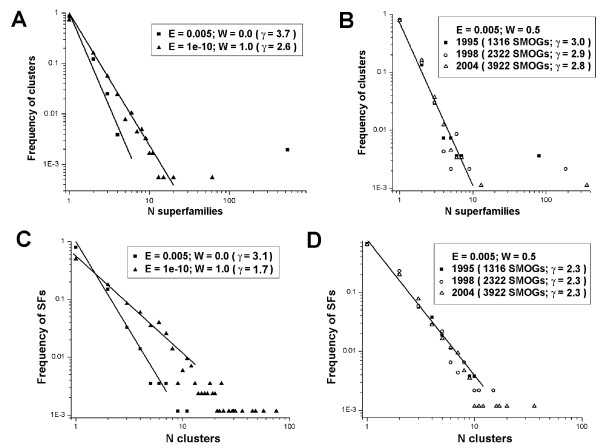
To make these assessments, we combine two approaches: (i) sequence analysis and homology-based structure prediction for proteins from complete genomes; and (ii) monitoring dynamics of the assigned structure set in time, with the accumulation of experimentally solved structures. In the Clusters of Orthologous Groups (COG) database, we map the growing population of structurally characterized domain families onto the network of sequence-based connections between domains. This mapping reveals a systematic bias suggesting that target families for structure determination tend to be located in highly populated areas of sequence space. In contrast, the subset of domains whose structure is initially inferred by SGI is similar to a random sample from the whole population. To accommodate for the observed bias, we propose a new non-parametric approach to the estimation of the total numbers of structural superfamilies and folds, which does not rely on a specific model of the sampling process. Based on dynamics of robust distribution-based parameters in the growing set of structure predictions, we estimate the total numbers of superfamilies and folds among soluble globular proteins in the COG database.
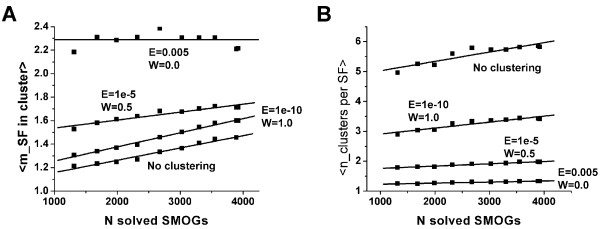
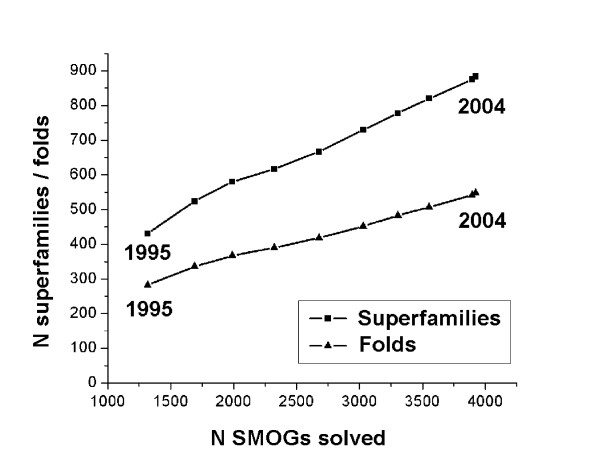
The set of currently solved protein structures allows for structure prediction in approximately a third of sequence-based domain families. The choice of targets for structure determination is biased towards domains with many sequence-based homologs. The growing SGI output in the future should further contribute to the reduction of this bias. The total number of structural superfamilies and folds in the COG database are estimated as approximately 4000 and approximately 1700. These numbers are respectively four and three times higher than the numbers of superfamilies and folds that can currently be assigned to COG proteins.
由于目前仅一小部分已知蛋白质家族具有三级结构,因此评估序列空间的哪些部分已进行结构表征非常重要。我们考虑其结构可通过与已解析结构的蛋白质的序列相似性来预测的蛋白质结构域,并解决以下问题。这些结构域是否代表所有序列家族的无偏随机样本?结构基因组计划(SGI)解析的目标是否提供这样的样本?可溶性球状结构域中基于结构的超家族和折叠的大致总数是多少?
为进行这些评估,我们结合了两种方法:(i)对来自完整基因组的蛋白质进行序列分析和基于同源性的结构预测;(ii)随着实验解析结构的积累,及时监测已分配结构集的动态变化。在直系同源群(COG)数据库中,我们将结构表征的结构域家族不断增长的群体映射到基于序列的结构域之间的连接网络上。这种映射揭示了一种系统性偏差,表明用于结构确定的目标家族往往位于序列空间中人口密集的区域。相比之下,最初由SGI推断其结构的结构域子集类似于来自整个人口的随机样本。为了适应观察到的偏差,我们提出了一种新的非参数方法来估计结构超家族和折叠的总数,该方法不依赖于采样过程的特定模型。基于不断增长的结构预测集中基于稳健分布的参数的动态变化,我们估计了COG数据库中可溶性球状蛋白质中超家族和折叠的总数。
当前解析的蛋白质结构集允许在大约三分之一基于序列的结构域家族中进行结构预测。结构确定目标的选择偏向于具有许多基于序列的同源物的结构域。未来不断增加的SGI产出应进一步有助于减少这种偏差。COG数据库中结构超家族和折叠的总数估计约为4000和约1700。这些数字分别比目前可分配给COG蛋白质的超家族和折叠的数量高出四倍和三倍。