Buck Institute for Age Research, Novato, California, United States of America.
PLoS One. 2007 May 9;2(5):e421. doi: 10.1371/journal.pone.0000421.
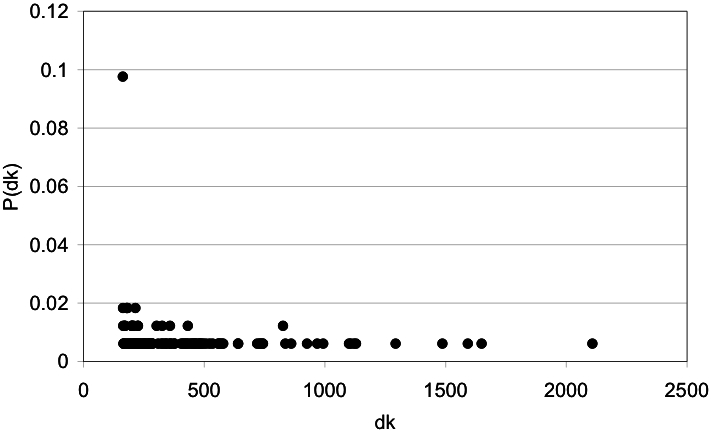
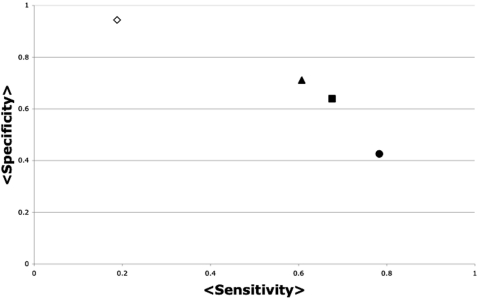
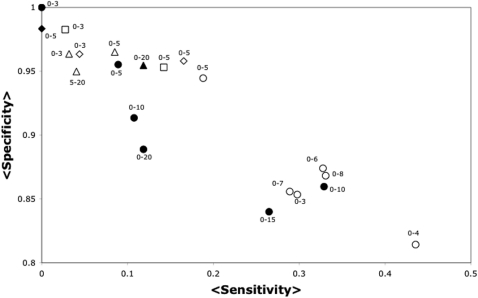
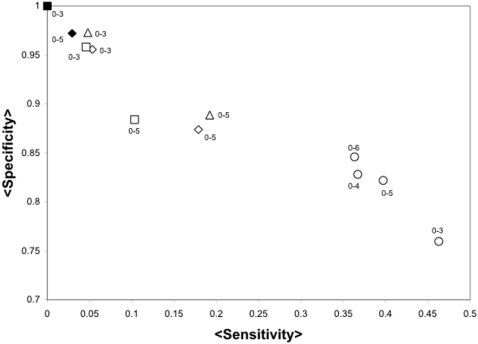
Despite the increasing number of published protein structures, and the fact that each protein's function relies on its three-dimensional structure, there is limited access to automatic programs used for the identification of critical residues from the protein structure, compared with those based on protein sequence. Here we present a new algorithm based on network analysis applied exclusively on protein structures to identify critical residues. Our results show that this method identifies critical residues for protein function with high reliability and improves automatic sequence-based approaches and previous network-based approaches. The reliability of the method depends on the conformational diversity screened for the protein of interest. We have designed a web site to give access to this software at http://bis.ifc.unam.mx/jamming/. In summary, a new method is presented that relates critical residues for protein function with the most traversed residues in networks derived from protein structures. A unique feature of the method is the inclusion of the conformational diversity of proteins in the prediction, thus reproducing a basic feature of the structure/function relationship of proteins.
尽管已发表的蛋白质结构数量不断增加,而且事实上每种蛋白质的功能都依赖于其三维结构,但与基于蛋白质序列的方法相比,用于从蛋白质结构中识别关键残基的自动程序的可及性有限。在这里,我们提出了一种新的算法,该算法基于网络分析,专门应用于蛋白质结构,以识别关键残基。我们的结果表明,该方法能够可靠地识别蛋白质功能的关键残基,并改进自动基于序列的方法和以前基于网络的方法。该方法的可靠性取决于为感兴趣的蛋白质筛选的构象多样性。我们设计了一个网站,可在 http://bis.ifc.unam.mx/jamming/ 上访问该软件。总之,提出了一种新的方法,将蛋白质功能的关键残基与从蛋白质结构中得出的网络中最常遍历的残基联系起来。该方法的一个独特特征是在预测中包含蛋白质的构象多样性,从而再现了蛋白质结构/功能关系的一个基本特征。