Roden Joseph C, King Brandon W, Trout Diane, Mortazavi Ali, Wold Barbara J, Hart Christopher E
Jet Propulsion Laboratory, California Institute of Technology, Pasadena, USA.
BMC Bioinformatics. 2006 Apr 7;7:194. doi: 10.1186/1471-2105-7-194.
There are many methods for analyzing microarray data that group together genes having similar patterns of expression over all conditions tested. However, in many instances the biologically important goal is to identify relatively small sets of genes that share coherent expression across only some conditions, rather than all or most conditions as required in traditional clustering; e.g. genes that are highly up-regulated and/or down-regulated similarly across only a subset of conditions. Equally important is the need to learn which conditions are the decisive ones in forming such gene sets of interest, and how they relate to diverse conditional covariates, such as disease diagnosis or prognosis.
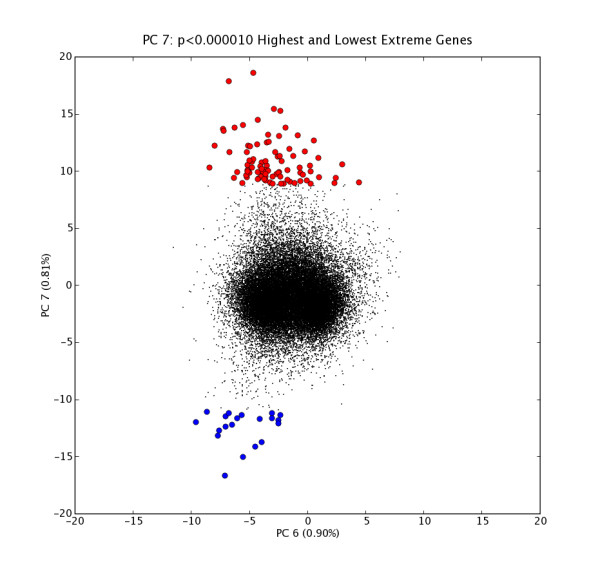
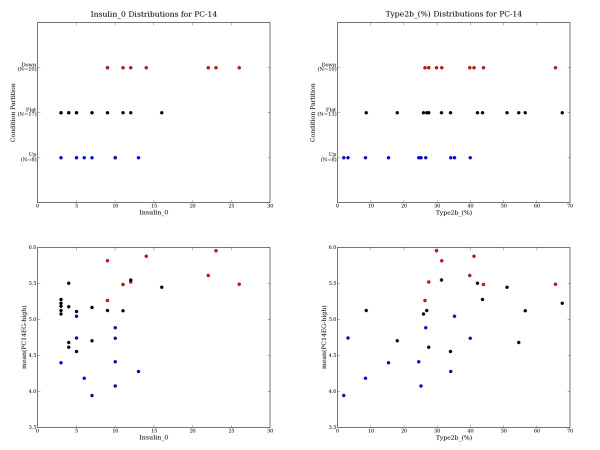
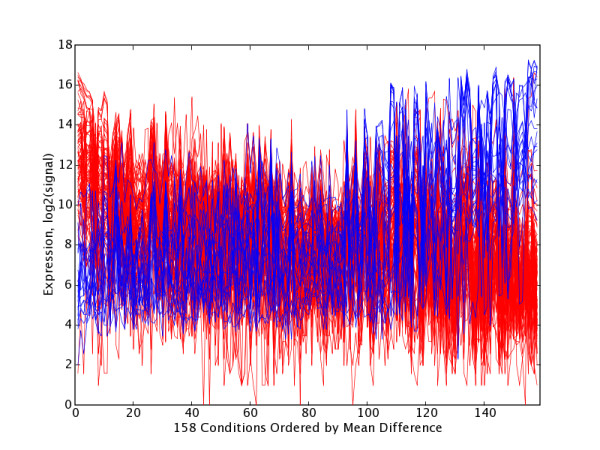
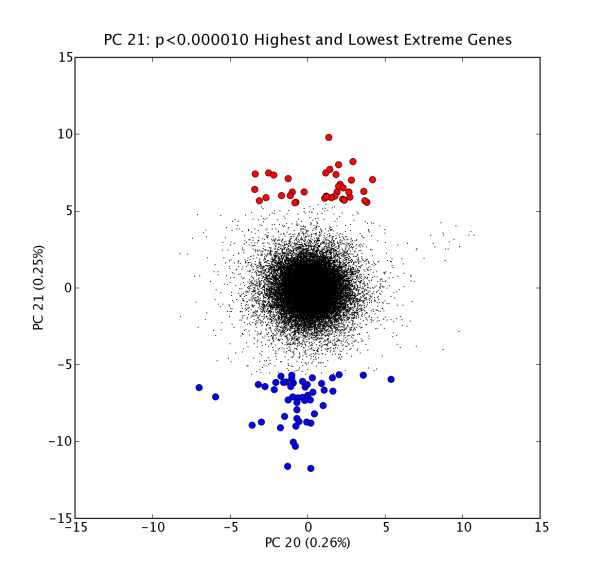
We present a method for automatically identifying such candidate sets of biologically relevant genes using a combination of principal components analysis and information theoretic metrics. To enable easy use of our methods, we have developed a data analysis package that facilitates visualization and subsequent data mining of the independent sources of significant variation present in gene microarray expression datasets (or in any other similarly structured high-dimensional dataset). We applied these tools to two public datasets, and highlight sets of genes most affected by specific subsets of conditions (e.g. tissues, treatments, samples, etc.). Statistically significant associations for highlighted gene sets were shown via global analysis for Gene Ontology term enrichment. Together with covariate associations, the tool provides a basis for building testable hypotheses about the biological or experimental causes of observed variation.
We provide an unsupervised data mining technique for diverse microarray expression datasets that is distinct from major methods now in routine use. In test uses, this method, based on publicly available gene annotations, appears to identify numerous sets of biologically relevant genes. It has proven especially valuable in instances where there are many diverse conditions (10's to hundreds of different tissues or cell types), a situation in which many clustering and ordering algorithms become problematic. This approach also shows promise in other topic domains such as multi-spectral imaging datasets.
有许多方法可用于分析微阵列数据,这些方法会将在所有测试条件下具有相似表达模式的基因归为一组。然而,在许多情况下,生物学上的重要目标是识别仅在某些条件下而非传统聚类所要求的所有或大多数条件下具有一致表达的相对较小的基因集;例如,仅在一部分条件下相似地上调或下调的基因。同样重要的是,需要了解哪些条件是形成此类感兴趣基因集的决定性条件,以及它们与各种条件协变量(如疾病诊断或预后)的关系。
我们提出了一种结合主成分分析和信息论指标自动识别此类生物学相关基因候选集的方法。为了便于使用我们的方法,我们开发了一个数据分析包,该包有助于对基因微阵列表达数据集(或任何其他类似结构的高维数据集)中存在的显著变异的独立来源进行可视化和后续数据挖掘。我们将这些工具应用于两个公共数据集,并突出显示受特定条件子集(如组织、处理、样本等)影响最大的基因集。通过对基因本体术语富集的全局分析显示了突出显示的基因集的统计学显著关联。连同协变量关联,该工具为建立关于观察到的变异的生物学或实验原因的可测试假设提供了基础。
我们为各种微阵列表达数据集提供了一种无监督数据挖掘技术,该技术与目前常规使用的主要方法不同。在测试使用中,基于公开可用基因注释的这种方法似乎可以识别许多生物学相关基因集。在存在许多不同条件(数十种到数百种不同组织或细胞类型)的情况下,这种方法已被证明特别有价值,在这种情况下,许多聚类和排序算法会出现问题。这种方法在其他主题领域(如多光谱成像数据集)也显示出前景。