Callegaro A, Spinelli R, Beltrame L, Bicciato S, Caristina L, Censuales S, De Bellis G, Battaglia C
Department of Chemical Process Engineering, University of Padua, Padua, Italy.
Nucleic Acids Res. 2006 Apr 14;34(7):e56. doi: 10.1093/nar/gkl185.
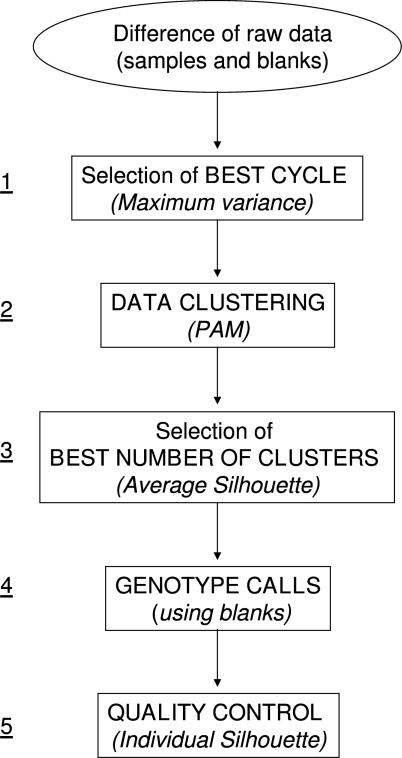
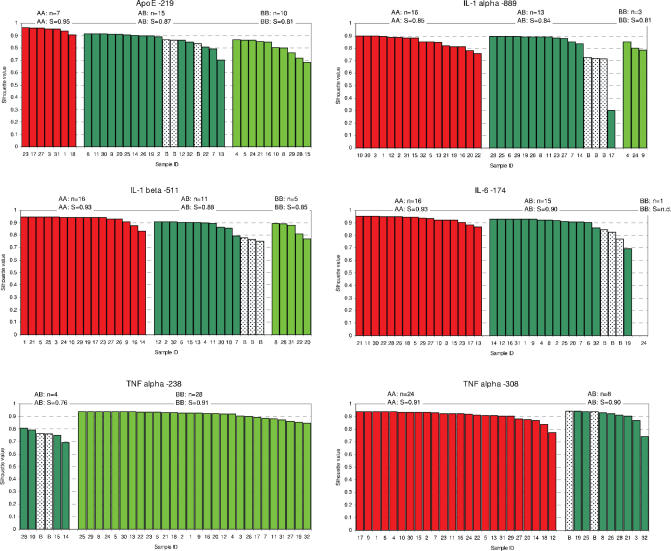
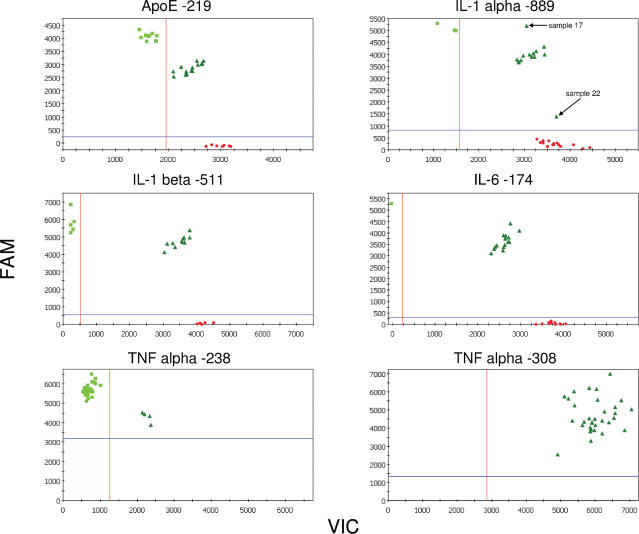
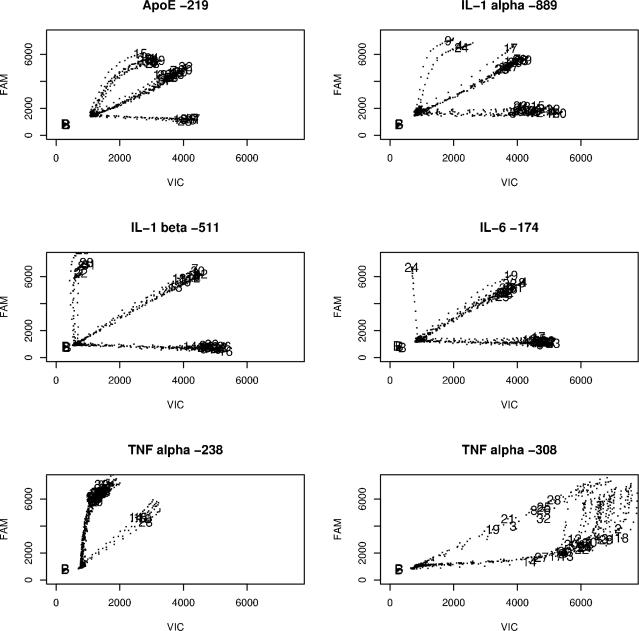
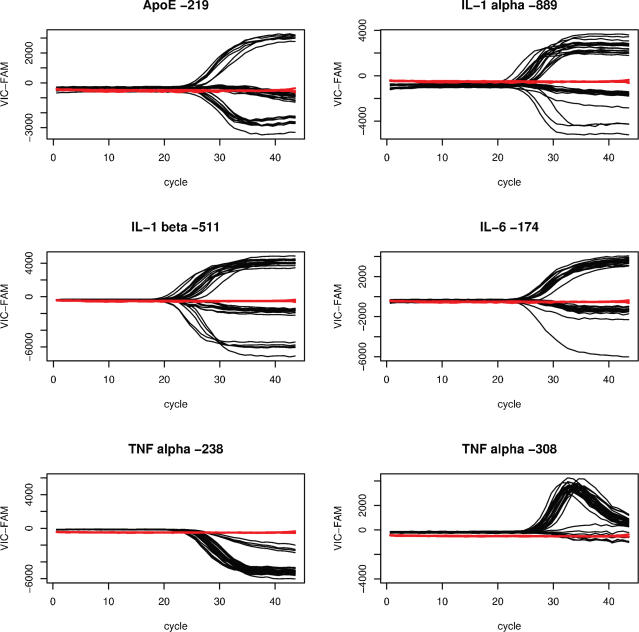
Single nucleotide polymorphisms (SNPs) are often determined using TaqMan real-time PCR assays (Applied Biosystems) and commercial software that assigns genotypes based on reporter probe signals at the end of amplification. Limitations to the large-scale application of this approach include the need for positive controls or operator intervention to set signal thresholds when one allele is rare. In the interest of optimizing real-time PCR genotyping, we developed an algorithm for automatic genotype calling based on the full course of real-time PCR data. Best cycle genotyping algorithm (BCGA), written in the open source language R, is based on the assumptions that classification depends on the time (cycle) of amplification and that it is possible to identify a best discriminating cycle for each SNP assay. The algorithm is unique in that it classifies samples according to the behavior of blanks (no DNA samples), which cluster with heterozygous samples. This method of classification eliminates the need for positive controls and permits accurate genotyping even in the absence of a genotype class, for example when one allele is rare. Here, we describe the algorithm and test its validity, compared to the standard end-point method and to DNA sequencing.
单核苷酸多态性(SNPs)通常使用TaqMan实时荧光定量PCR检测法(应用生物系统公司)和商业软件来确定,该软件会根据扩增结束时报告探针的信号来确定基因型。这种方法大规模应用的局限性包括,当一个等位基因罕见时,需要阳性对照或操作人员干预来设置信号阈值。为了优化实时荧光定量PCR基因分型,我们基于实时荧光定量PCR数据的全过程开发了一种自动基因型判读算法。最佳循环基因分型算法(BCGA)用开源语言R编写,基于这样的假设:分类取决于扩增时间(循环数),并且有可能为每个SNP检测确定一个最佳区分循环。该算法的独特之处在于,它根据空白样本(无DNA样本)的行为对样本进行分类,空白样本与杂合样本聚集在一起。这种分类方法无需阳性对照,即使在没有基因型类别的情况下,例如当一个等位基因罕见时,也能进行准确的基因分型。在此,我们描述该算法,并将其与标准终点法和DNA测序法相比较,测试其有效性。