Dobson Richard J, Munroe Patricia B, Caulfield Mark J, Saqi Mansoor As
Clinical Pharmacology, The William Harvey Research Institute, Bart's and the London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK.
BMC Bioinformatics. 2006 Apr 21;7:217. doi: 10.1186/1471-2105-7-217.
There has been an explosion in the number of single nucleotide polymorphisms (SNPs) within public databases. In this study we focused on non-synonymous protein coding single nucleotide polymorphisms (nsSNPs), some associated with disease and others which are thought to be neutral. We describe the distribution of both types of nsSNPs using structural and sequence based features and assess the relative value of these attributes as predictors of function using machine learning methods. We also address the common problem of balance within machine learning methods and show the effect of imbalance on nsSNP function prediction. We show that nsSNP function prediction can be significantly improved by 100% undersampling of the majority class. The learnt rules were then applied to make predictions of function on all nsSNPs within Ensembl.
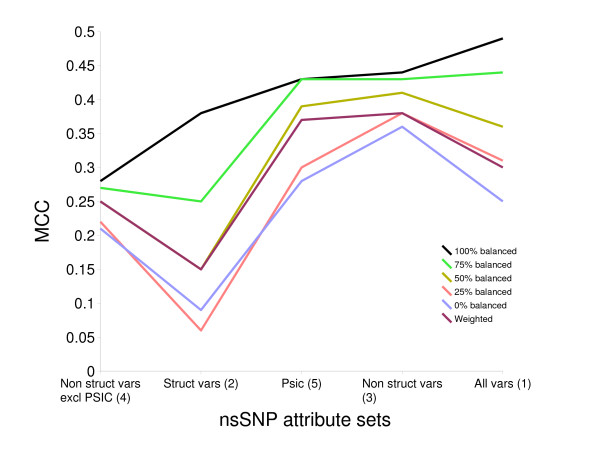
The measure of prediction success is greatly affected by the level of imbalance in the training dataset. We found the balanced dataset that included all attributes produced the best prediction. The performance as measured by the Matthews correlation coefficient (MCC) varied between 0.49 and 0.25 depending on the imbalance. As previously observed, the degree of sequence conservation at the nsSNP position is the single most useful attribute. In addition to conservation, structural predictions made using a balanced dataset can be of value.
The predictions for all nsSNPs within Ensembl, based on a balanced dataset using all attributes, are available as a DAS annotation. Instructions for adding the track to Ensembl are at http://www.brightstudy.ac.uk/das_help.html.
公共数据库中单个核苷酸多态性(SNP)的数量呈爆炸式增长。在本研究中,我们聚焦于非同义蛋白质编码单核苷酸多态性(nsSNP),其中一些与疾病相关,另一些则被认为是中性的。我们使用基于结构和序列的特征描述了这两种类型的nsSNP的分布,并使用机器学习方法评估这些属性作为功能预测指标的相对价值。我们还解决了机器学习方法中常见的平衡问题,并展示了不平衡对nsSNP功能预测的影响。我们表明,通过对多数类进行100%欠采样,nsSNP功能预测可得到显著改善。然后将学习到的规则应用于对Ensembl中所有nsSNP的功能进行预测。
预测成功的度量受训练数据集不平衡程度极大影响。我们发现包含所有属性的平衡数据集产生了最佳预测。根据不平衡程度,由马修斯相关系数(MCC)衡量的性能在0.49至0.25之间变化。如先前观察到的,nsSNP位置处的序列保守程度是最有用的单个属性。除了保守性之外,使用平衡数据集进行的结构预测也可能有价值。
基于使用所有属性的平衡数据集对Ensembl中所有nsSNP的预测以DAS注释形式提供。将该轨迹添加到Ensembl的说明可在http://www.brightstudy.ac.uk/das_help.html获取。