Tian Jian, Wu Ningfeng, Guo Xuexia, Guo Jun, Zhang Juhua, Fan Yunliu
Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing 100081, China.
BMC Bioinformatics. 2007 Nov 16;8:450. doi: 10.1186/1471-2105-8-450.
Human genetic variations primarily result from single nucleotide polymorphisms (SNPs) that occur approximately every 1000 bases in the overall human population. The non-synonymous SNPs (nsSNPs) that lead to amino acid changes in the protein product may account for nearly half of the known genetic variations linked to inherited human diseases. One of the key problems of medical genetics today is to identify nsSNPs that underlie disease-related phenotypes in humans. As such, the development of computational tools that can identify such nsSNPs would enhance our understanding of genetic diseases and help predict the disease.
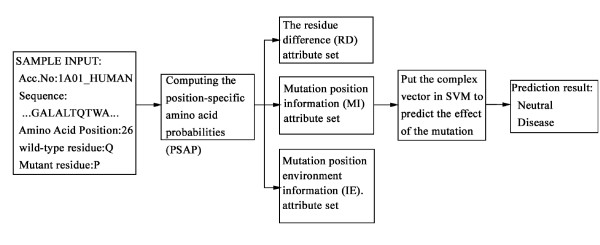
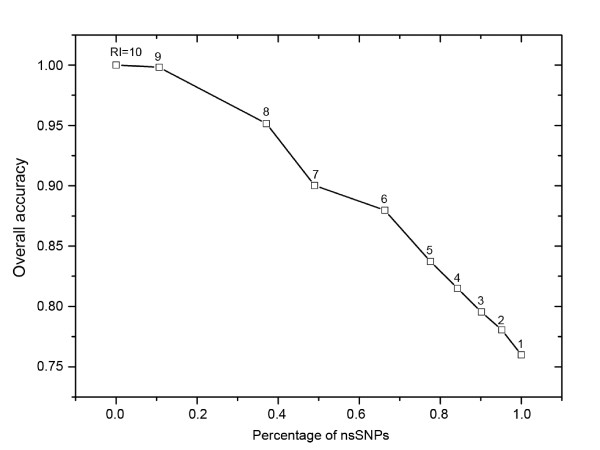
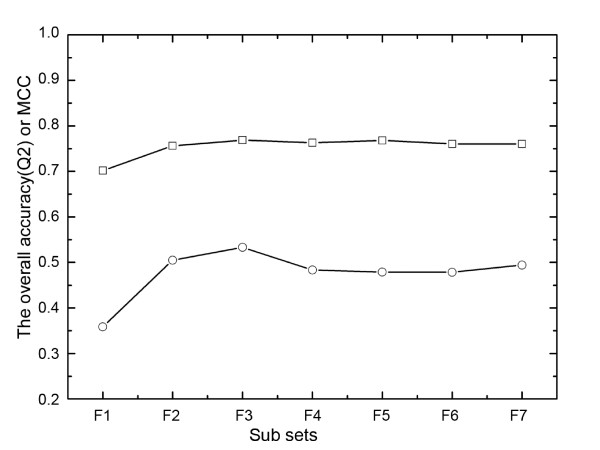
We propose a method, named Parepro (Predicting the amino acid replacement probability), to identify nsSNPs having either deleterious or neutral effects on the resulting protein function. Two independent datasets, HumVar and NewHumVar, taken from the PhD-SNP server, were applied to train the model and test the robustness of Parepro. Using a 20-fold cross validation test on the HumVar dataset, Parepro achieved a Matthews correlation coefficient (MCC) of 50% and an overall accuracy (Q2) of 76%, both of which were higher than those predicted by the methods, such as PolyPhen, SIFT, and HydridMeth. Further analysis on an additional dataset (NewHumVar) using Parepro yielded similar results.
The performance of Parepro indicates that it is a powerful tool for predicting the effect of nsSNPs on protein function and would be useful for large-scale analysis of genomic nsSNP data.
人类遗传变异主要源于单核苷酸多态性(SNP),在整个人口中大约每1000个碱基就会出现一次。导致蛋白质产物中氨基酸变化的非同义SNP(nsSNP)可能占已知与人类遗传性疾病相关的遗传变异的近一半。当今医学遗传学的关键问题之一是识别导致人类疾病相关表型的nsSNP。因此,开发能够识别此类nsSNP的计算工具将增进我们对遗传疾病的理解,并有助于预测疾病。
我们提出了一种名为Parepro(预测氨基酸替代概率)的方法,以识别对所得蛋白质功能具有有害或中性影响的nsSNP。从PhD-SNP服务器获取的两个独立数据集HumVar和NewHumVar用于训练模型并测试Parepro的稳健性。在HumVar数据集上使用20倍交叉验证测试,Parepro的马修斯相关系数(MCC)达到50%,总体准确率(Q2)达到76%,两者均高于PolyPhen、SIFT和HydridMeth等方法的预测值。使用Parepro对另一个数据集(NewHumVar)进行的进一步分析得出了类似的结果。
Parepro的性能表明它是预测nsSNP对蛋白质功能影响的强大工具,将有助于对基因组nsSNP数据进行大规模分析。