Tiemann-Boege Irene, Calabrese Peter, Cochran David M, Sokol Rebecca, Arnheim Norman
Molecular and Computational Biology Program, University of Southern California, Los Angeles, California, USA.
PLoS Genet. 2006 May;2(5):e70. doi: 10.1371/journal.pgen.0020070. Epub 2006 May 5.
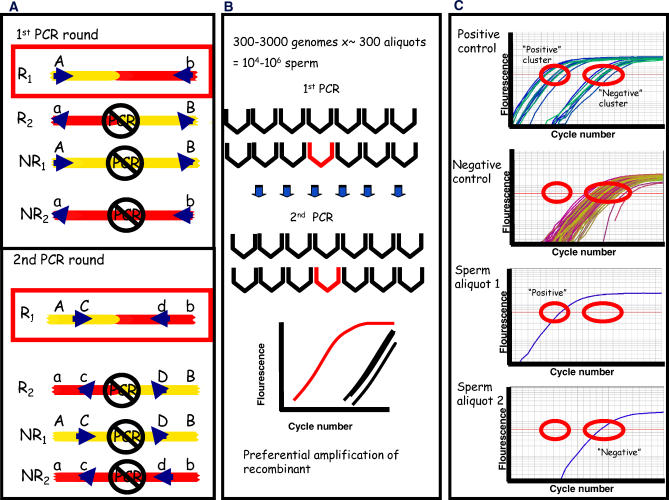
For decades, classical crossover studies and linkage disequilibrium (LD) analysis of genomic regions suggested that human meiotic crossovers may not be randomly distributed along chromosomes but are focused instead in "hot spots." Recent sperm typing studies provided data at very high resolution and accuracy that defined the physical limits of a number of hot spots. The data were also used to test whether patterns of LD can predict hot spot locations. These sperm typing studies focused on several small regions of the genome already known or suspected of containing a hot spot based on the presence of LD breakdown or previous experimental evidence of hot spot activity. Comparable data on target regions not specifically chosen using these two criteria is lacking but is needed to make an unbiased test of whether LD data alone can accurately predict active hot spots. We used sperm typing to estimate recombination in 17 almost contiguous ~5 kb intervals spanning 103 kb of human Chromosome 21. We found two intervals that contained new hot spots. The comparison of our data with recombination rates predicted by statistical analyses of LD showed that, overall, the two datasets corresponded well, except for one predicted hot spot that showed little crossing over. This study doubles the experimental data on recombination in men at the highest resolution and accuracy and supports the emerging genome-wide picture that recombination is localized in small regions separated by cold areas. Detailed study of one of the new hot spots revealed a sperm donor with a decrease in recombination intensity at the canonical recombination site but an increase in crossover activity nearby. This unique finding suggests that the position and intensity of hot spots may evolve by means of a concerted mechanism that maintains the overall recombination intensity in the region.
几十年来,对基因组区域的经典交叉研究和连锁不平衡(LD)分析表明,人类减数分裂交叉可能并非沿染色体随机分布,而是集中在“热点”区域。最近的精子分型研究提供了高分辨率和高精度的数据,确定了多个热点的物理界限。这些数据还用于测试LD模式是否能够预测热点位置。这些精子分型研究聚焦于基因组中几个已知或疑似含有热点的小区域,这些区域是基于LD断裂的存在或先前热点活性的实验证据确定的。目前缺乏关于未根据这两个标准专门选择的目标区域的可比数据,但这对于公正测试仅靠LD数据能否准确预测活跃热点是必要的。我们利用精子分型来估计人类21号染色体上17个几乎连续的约5kb区间(跨度为103kb)的重组情况。我们发现了两个包含新热点的区间。将我们的数据与通过LD统计分析预测的重组率进行比较,结果表明,总体而言,这两个数据集吻合得很好,只是有一个预测热点的交叉很少。这项研究将男性重组的实验数据在最高分辨率和精度上增加了一倍,并支持了新出现的全基因组图景,即重组定位于由冷区隔开的小区域。对其中一个新热点的详细研究发现,一名精子供体在典型重组位点的重组强度降低,但附近的交叉活性增加。这一独特发现表明,热点的位置和强度可能通过一种协调机制演变,该机制维持了该区域的总体重组强度。