Department of Applied Statistics, Johannes Kepler University Linz, Linz, Austria.
Institute of Biophysics, Johannes Kepler University Linz, Linz, Austria.
Mol Ecol Resour. 2019 May;19(3):623-638. doi: 10.1111/1755-0998.12994. Epub 2019 Apr 4.
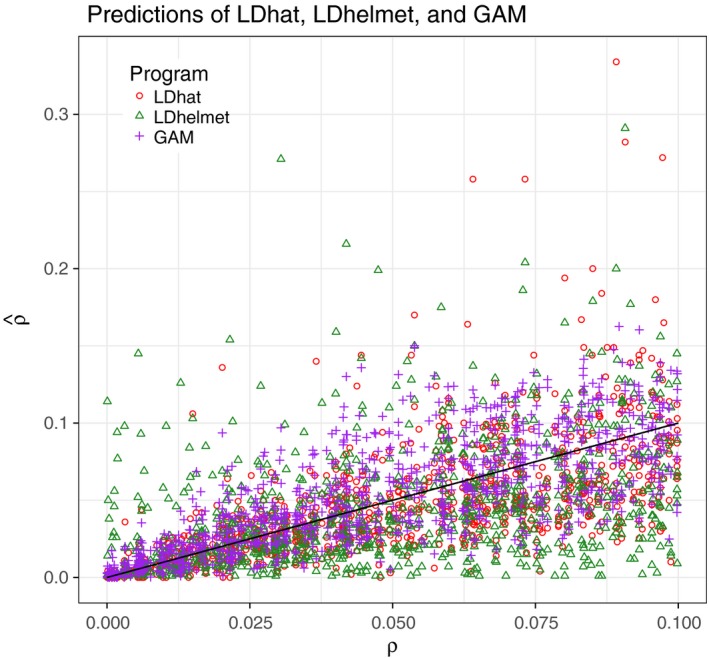
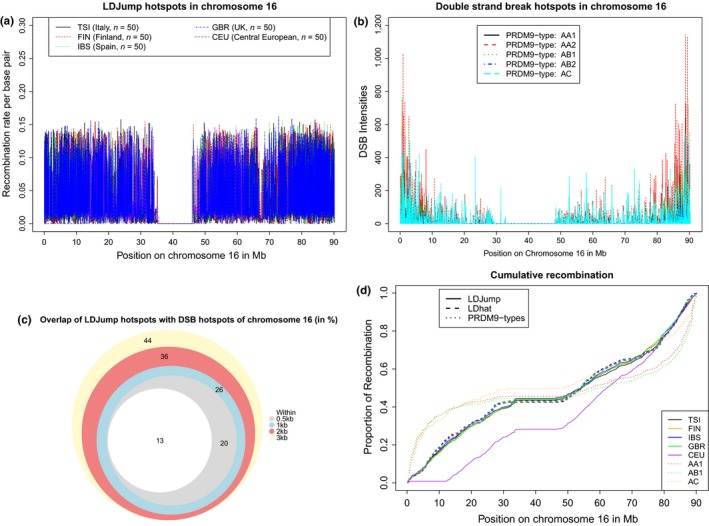
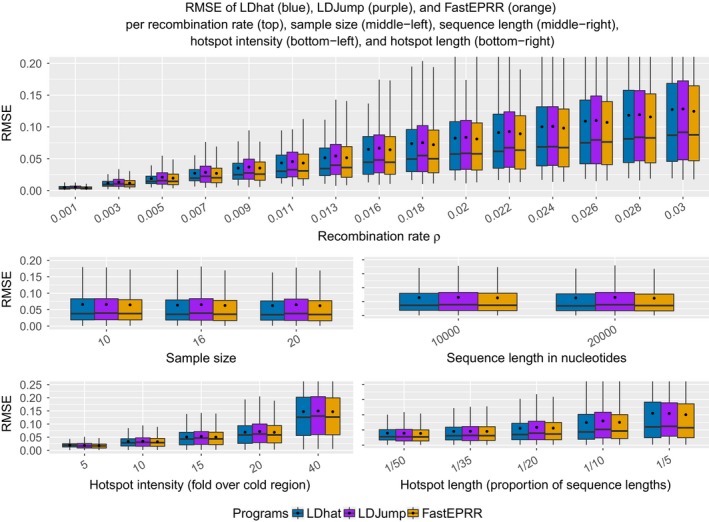
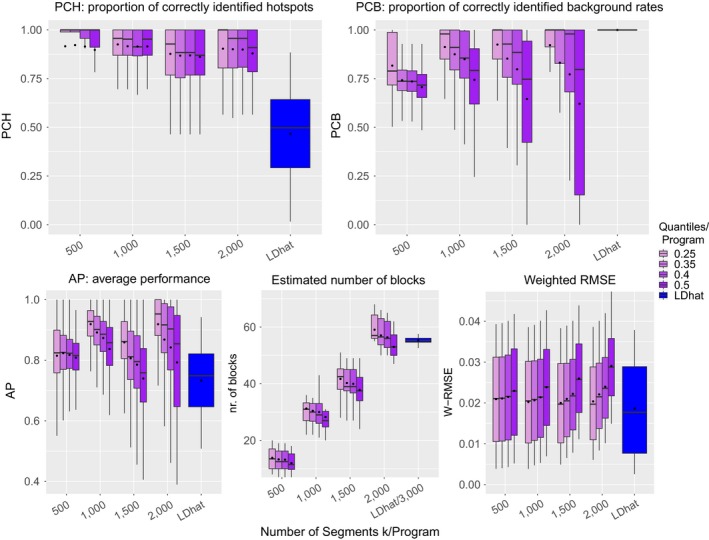
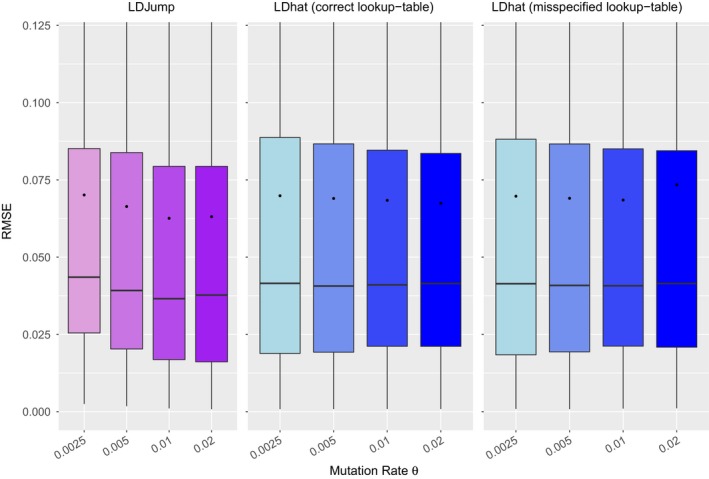
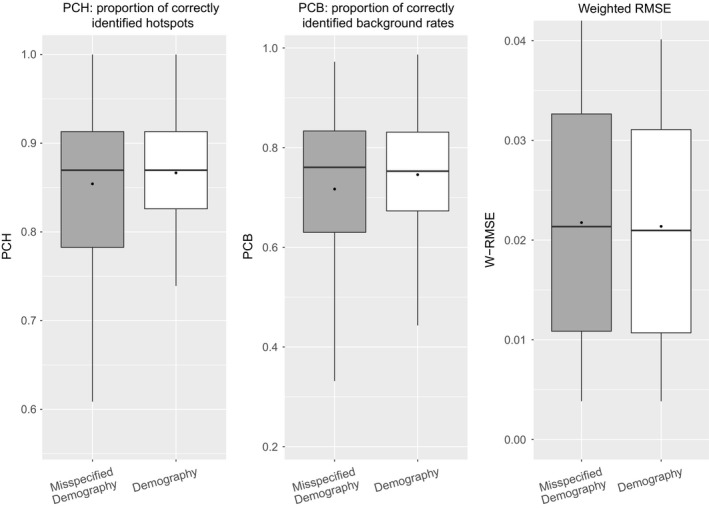
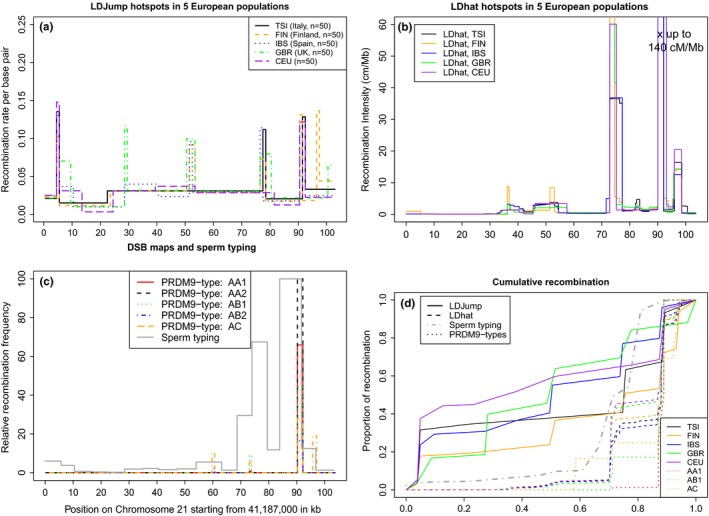
As recombination plays an important role in evolution, its estimation and the identification of hotspot positions is of considerable interest. We propose a novel approach for estimating population recombination rates based on genotyping or sequence data that involves a sequential multiscale change point estimator. Our method also permits demography to be taken into account. It uses several summary statistics within a regression model fitted on suitable scenarios. Our proposed method is accurate, computationally fast, and provides a parsimonious solution by ensuring a type I error control against too many changes in the recombination rate. An application to human genome data suggests a good congruence between our estimated and experimentally identified hotspots. Our method is implemented in the R-package LDJump, which is freely available at https://github.com/PhHermann/LDJump.
由于重组在进化中起着重要作用,因此估计重组率和识别热点位置具有相当大的意义。我们提出了一种基于基因分型或序列数据的估计群体重组率的新方法,该方法涉及顺序多尺度变点估计器。我们的方法还允许考虑人口统计学因素。它在回归模型中使用了几个汇总统计量,并在合适的场景中进行拟合。我们提出的方法准确、计算速度快,并且通过确保针对重组率的过多变化进行 I 型错误控制,提供了一种简洁的解决方案。对人类基因组数据的应用表明,我们估计的热点与实验确定的热点之间具有很好的一致性。我们的方法在 R 包 LDJump 中实现,该包可在 https://github.com/PhHermann/LDJump 上免费获得。