Tanay Amos, Steinfeld Israel, Kupiec Martin, Shamir Ron
School of Computer Science, Tel Aviv University, Tel Aviv, Israel.
Mol Syst Biol. 2005;1:2005.0002. doi: 10.1038/msb4100005. Epub 2005 Mar 29.
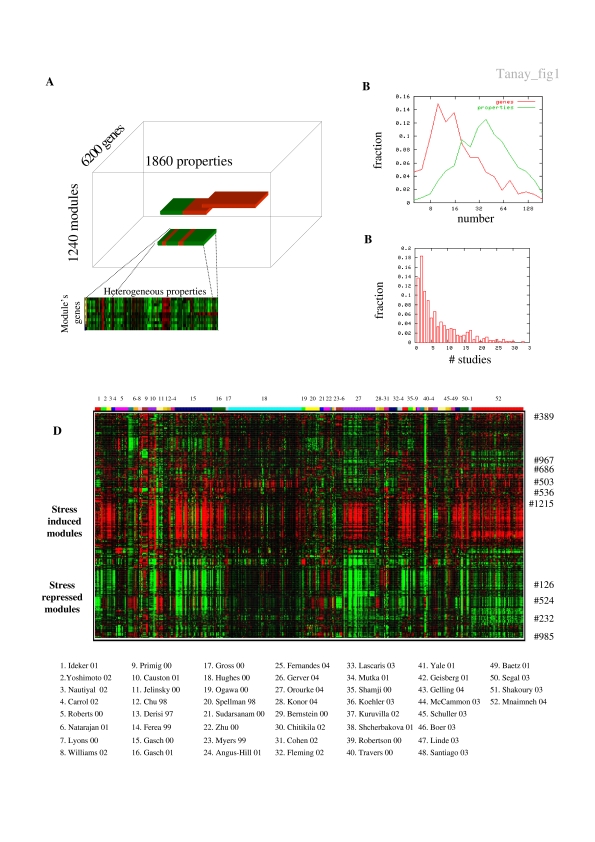
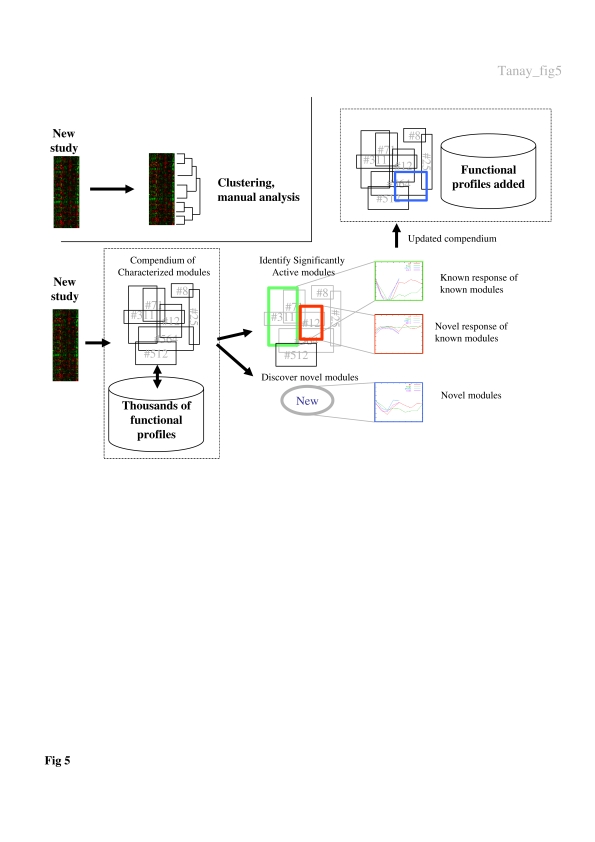
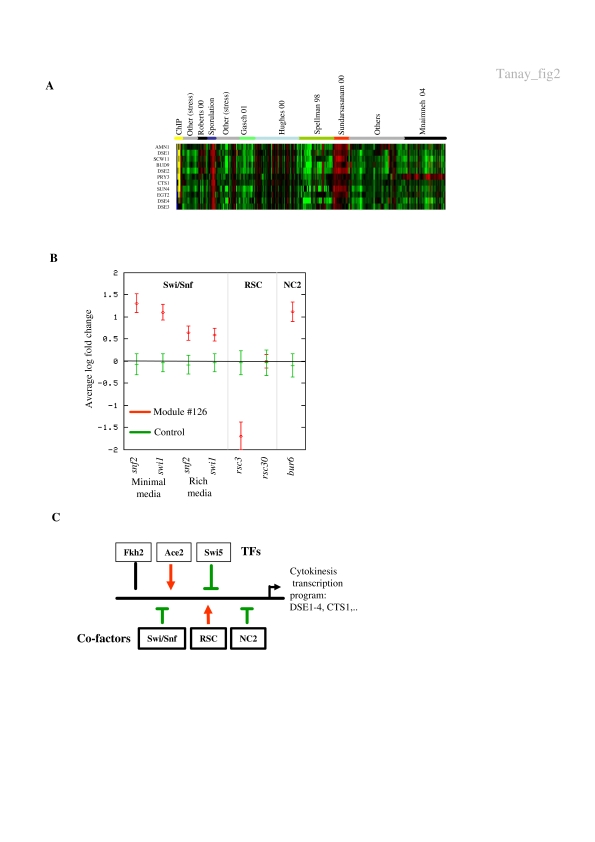
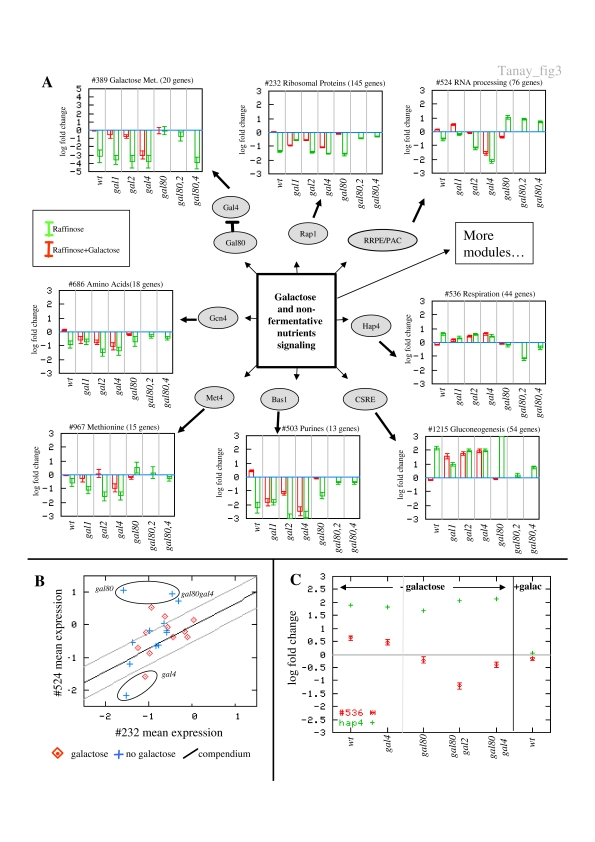
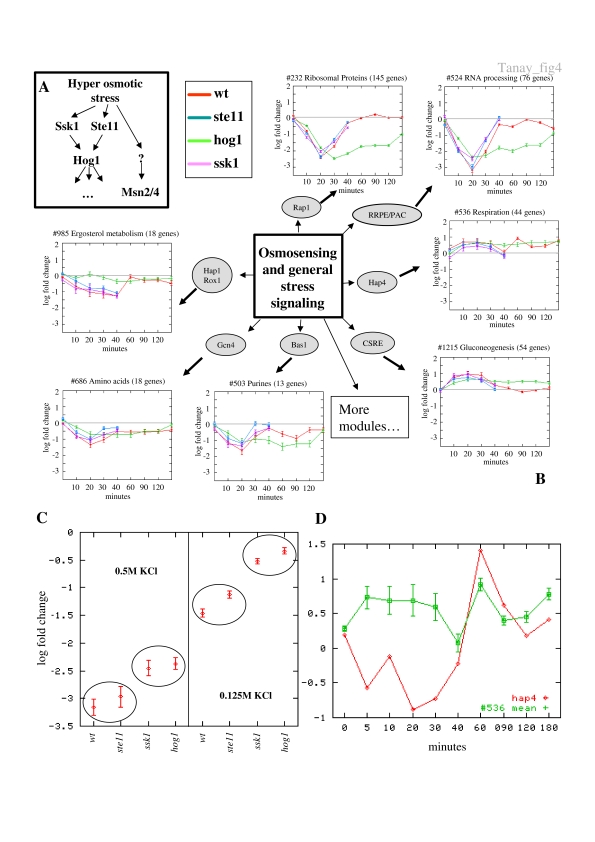
Biological systems are orchestrated by heterogeneous regulatory programs that control complex processes and adapt to a dynamic environment. Recent advances in high-throughput experimental methods provide genome-wide perspectives on such regulatory programs. A considerable amount of data on the behavior of model systems in a variety of conditions is rapidly accumulating. Still, the dominant paradigm is to analyze new genome-wide experiments separately from any other extant data, for example, by clustering the new data alone. Here we introduce a new methodology for analyzing the results of a new functional genomic study vis-à-vis a large compendium of previously published results from heterogeneous experimental techniques. We demonstrate our methodology on Saccharomyces cerevisiae, using a compendium of some 2000 experiments from 60 different publications. Most importantly, we show how the integrated analysis reveals unexpected connections among biological processes, and differentiates between novel and known effects in the analyzed experiments. Such characterization is impossible when new data sets are studied in isolation. Our results exemplify the power of the integrative approach in the analysis of genomic scale data sets and call for a paradigm shift in their study.
生物系统由控制复杂过程并适应动态环境的异质调控程序精心编排。高通量实验方法的最新进展为这类调控程序提供了全基因组视角。关于模型系统在各种条件下行为的大量数据正在迅速积累。然而,主流范式是将新的全基因组实验与任何其他现有数据分开分析,例如,仅对新数据进行聚类。在此,我们引入一种新方法,用于相对于大量来自异质实验技术的先前发表结果的汇编,分析新的功能基因组学研究结果。我们使用来自60个不同出版物的约2000个实验的汇编,在酿酒酵母上展示了我们的方法。最重要的是,我们展示了综合分析如何揭示生物过程之间意想不到的联系,并区分分析实验中的新效应和已知效应。当单独研究新数据集时,这种表征是不可能的。我们的结果例证了整合方法在基因组规模数据集分析中的强大作用,并呼吁在其研究中进行范式转变。