Davies Matthew N, Toseland Christopher P, Moss David S, Flower Darren R
Edward Jenner Institute for Vaccine Research, Compton, Berkshire, RG20 7NN, UK.
BMC Biochem. 2006 Jun 2;7:18. doi: 10.1186/1471-2091-7-18.
pKa values are a measure of the protonation of ionizable groups in proteins. Ionizable groups are involved in intra-protein, protein-solvent and protein-ligand interactions as well as solubility, protein folding and catalytic activity. The pKa shift of a group from its intrinsic value is determined by the perturbation of the residue by the environment and can be calculated from three-dimensional structural data.
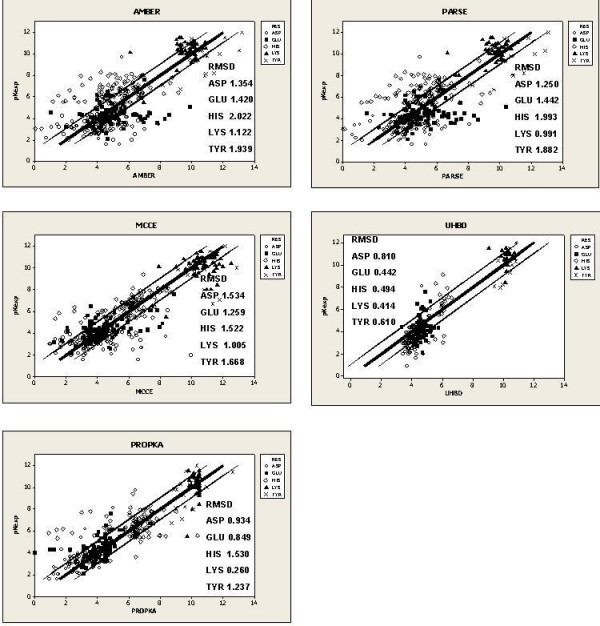
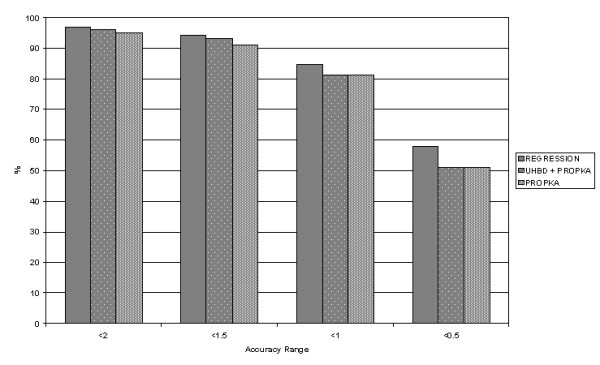
Here we use a large dataset of experimentally-determined pKas to analyse the performance of different prediction techniques. Our work provides a benchmark of available software implementations: MCCE, MEAD, PROPKA and UHBD. Combinatorial and regression analysis is also used in an attempt to find a consensus approach towards pKa prediction. The tendency of individual programs to over- or underpredict the pKa value is related to the underlying methodology of the individual programs.
Overall, PROPKA is more accurate than the other three programs. Key to developing accurate predictive software will be a complete sampling of conformations accessible to protein structures.
pKa值是衡量蛋白质中可电离基团质子化程度的指标。可电离基团参与蛋白质内部、蛋白质与溶剂以及蛋白质与配体的相互作用,同时也与溶解度、蛋白质折叠和催化活性有关。基团相对于其固有值的pKa偏移由环境对残基的扰动决定,并且可以从三维结构数据计算得出。
在此,我们使用一个由实验测定的pKa组成的大型数据集来分析不同预测技术的性能。我们的工作提供了现有软件实现的基准:MCCE、MEAD、PROPKA和UHBD。还使用了组合分析和回归分析,试图找到一种pKa预测的共识方法。各个程序对pKa值预测过高或过低的倾向与各个程序的基础方法有关。
总体而言,PROPKA比其他三个程序更准确。开发准确预测软件的关键将是对蛋白质结构可及的构象进行完整采样。