Schloss Patrick D, Handelsman Jo
Department of Plant Pathology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
PLoS Comput Biol. 2006 Jul 21;2(7):e92. doi: 10.1371/journal.pcbi.0020092. Epub 2006 Jun 5.
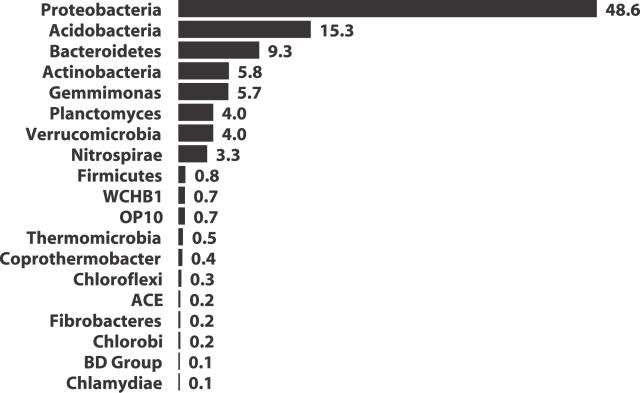
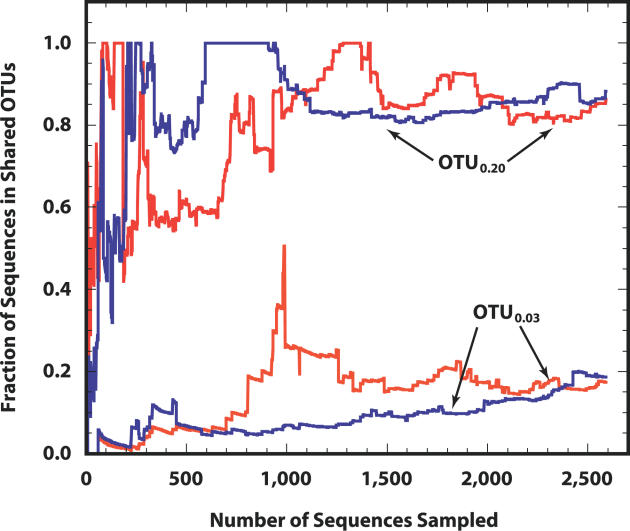
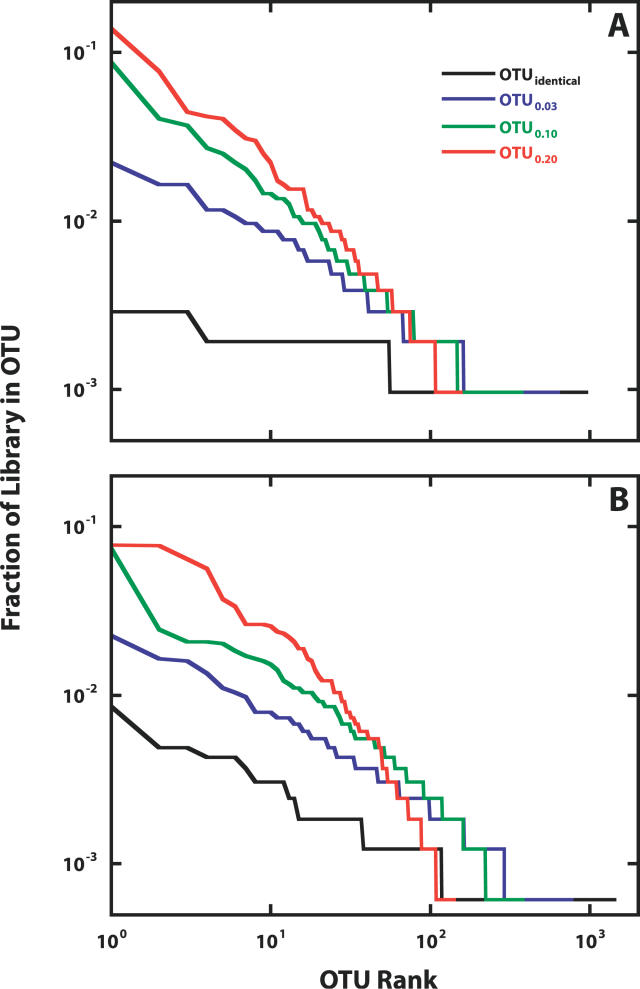
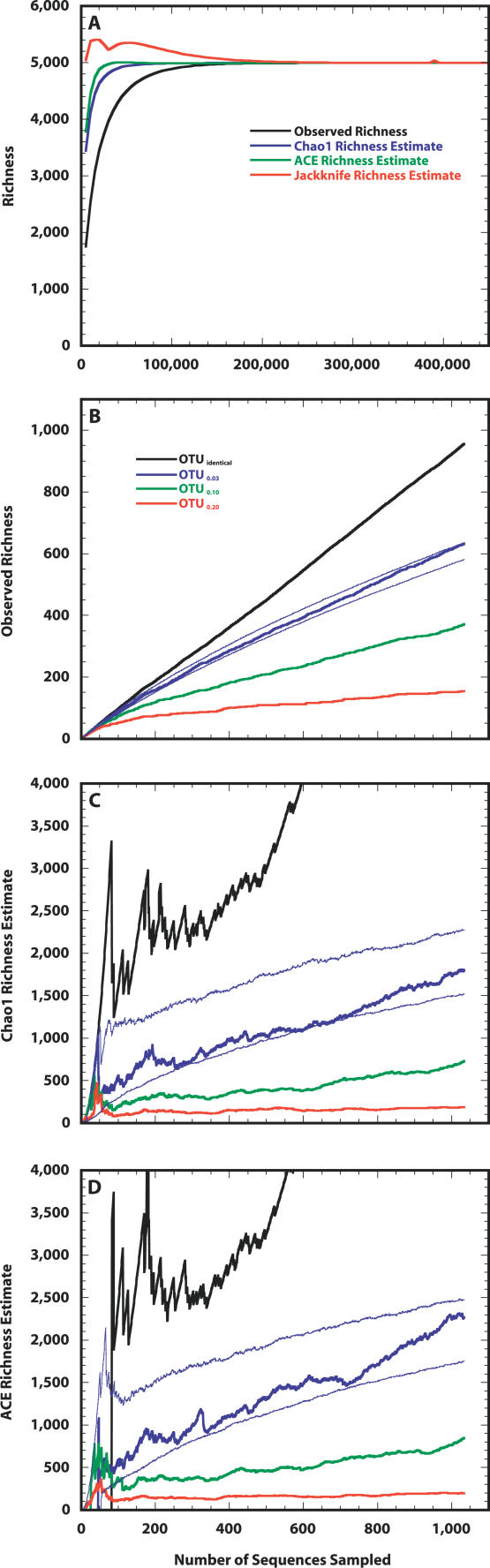
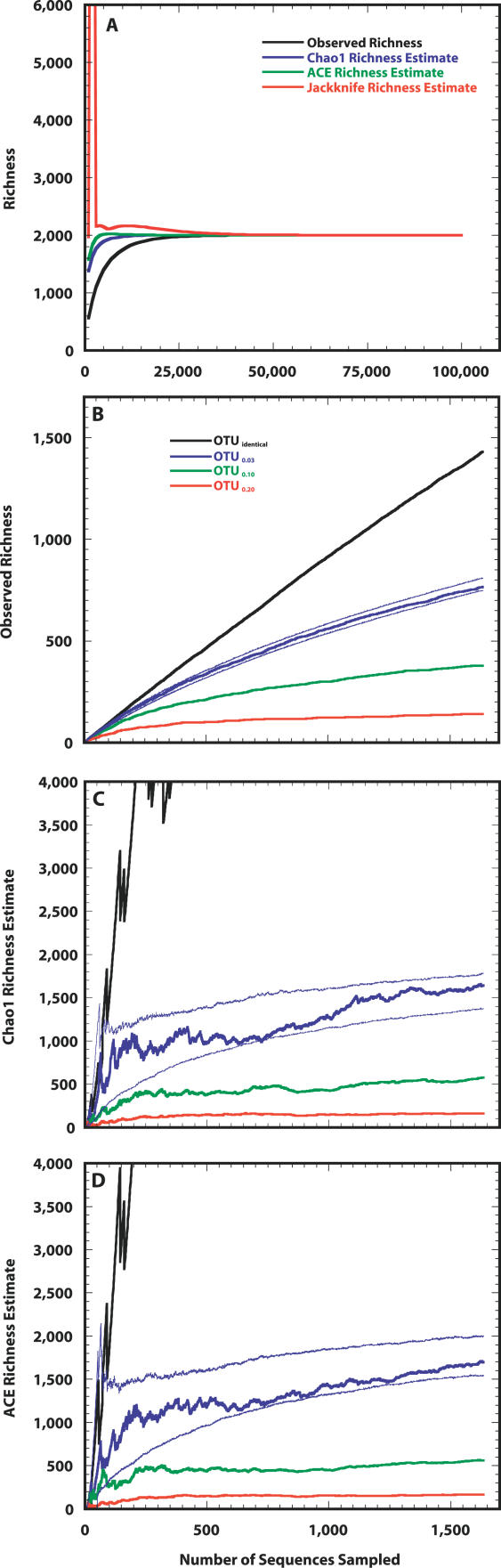
For more than a century, microbiologists have sought to determine the species richness of bacteria in soil, but the extreme complexity and unknown structure of soil microbial communities have obscured the answer. We developed a statistical model that makes the problem of estimating richness statistically accessible by evaluating the characteristics of samples drawn from simulated communities with parametric community distributions. We identified simulated communities with rank-abundance distributions that followed a truncated lognormal distribution whose samples resembled the structure of 16S rRNA gene sequence collections made using Alaskan and Minnesotan soils. The simulated communities constructed based on the distribution of 16S rRNA gene sequences sampled from the Alaskan and Minnesotan soils had a richness of 5,000 and 2,000 operational taxonomic units (OTUs), respectively, where an OTU represents a collection of sequences not more than 3% distant from each other. To sample each of these OTUs in the Alaskan 16S rRNA gene library at least twice, 480,000 sequences would be required; however, to estimate the richness of the simulated communities using nonparametric richness estimators would require only 18,000 sequences. Quantifying the richness of complex environments such as soil is an important step in building an ecological framework. We have shown that generating sufficient sequence data to do so requires less sequencing effort than completely sequencing a bacterial genome.
一个多世纪以来,微生物学家一直试图确定土壤中细菌的物种丰富度,但土壤微生物群落的极端复杂性和未知结构掩盖了答案。我们开发了一种统计模型,通过评估从具有参数化群落分布的模拟群落中抽取的样本特征,使估计丰富度的问题在统计上变得可行。我们确定了具有秩-丰度分布的模拟群落,其遵循截断对数正态分布,其样本类似于使用阿拉斯加和明尼苏达土壤制作的16S rRNA基因序列集合的结构。基于从阿拉斯加和明尼苏达土壤中采样的16S rRNA基因序列分布构建的模拟群落,其丰富度分别为5000个和2000个操作分类单元(OTU),其中一个OTU代表彼此距离不超过3%的序列集合。要在阿拉斯加16S rRNA基因文库中对每个这些OTU至少采样两次,需要480000个序列;然而,使用非参数丰富度估计器来估计模拟群落的丰富度仅需要18000个序列。量化土壤等复杂环境的丰富度是构建生态框架的重要一步。我们已经表明,为此生成足够的序列数据所需的测序工作量比完全测序一个细菌基因组要少。