Chardot Christophe
Service de chirurgie pédiatrique, Hôpital Cantonal Universitaire de Genève, Rue Willi Donzé 6, CH 1205 Geneve, Switzerland.
Orphanet J Rare Dis. 2006 Jul 26;1:28. doi: 10.1186/1750-1172-1-28.
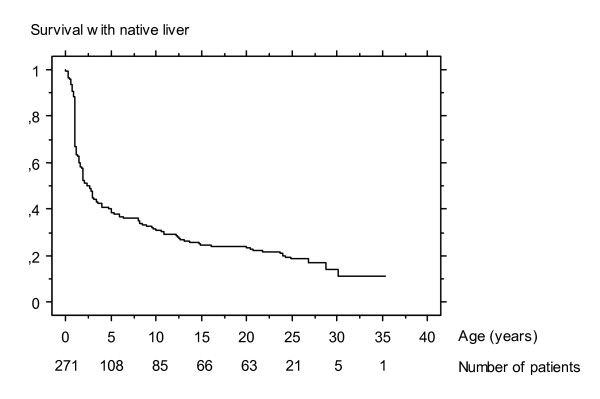
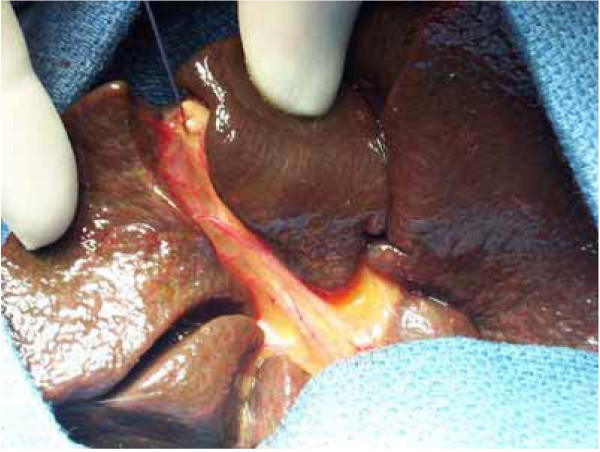
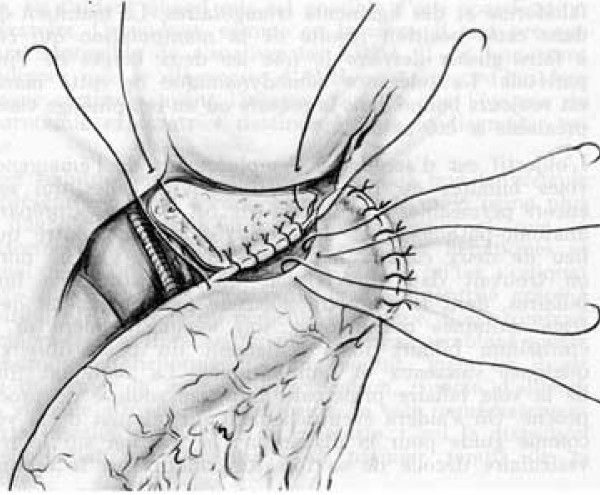
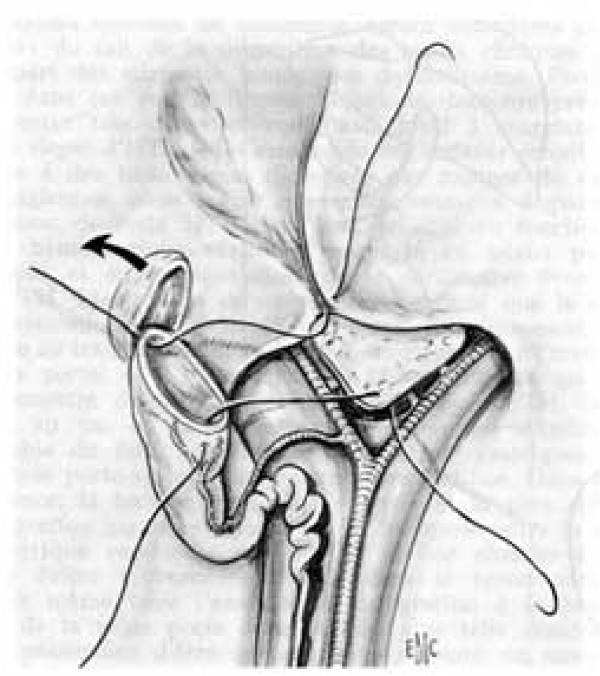
Biliary atresia (BA) is a rare disease characterised by a biliary obstruction of unknown origin that presents in the neonatal period. It is the most frequent surgical cause of cholestatic jaundice in this age group. BA occurs in approximately 1/18,000 live births in Western Europe. In the world, the reported incidence varies from 5/100,000 to 32/100,000 live births, and is highest in Asia and the Pacific region. Females are affected slightly more often than males. The common histopathological picture is one of inflammatory damage to the intra- and extrahepatic bile ducts with sclerosis and narrowing or even obliteration of the biliary tree. Untreated, this condition leads to cirrhosis and death within the first years of life. BA is not known to be a hereditary condition. No primary medical treatment is relevant for the management of BA. Once BA suspected, surgical intervention (Kasai portoenterostomy) should be performed as soon as possible as operations performed early in life is more likely to be successful. Liver transplantation may be needed later if the Kasai operation fails to restore the biliary flow or if cirrhotic complications occur. At present, approximately 90% of BA patients survive and the majority have normal quality of life.
胆道闭锁(BA)是一种罕见疾病,其特征为新生儿期出现病因不明的胆道梗阻。它是该年龄组胆汁淤积性黄疸最常见的手术病因。在西欧,BA的发病率约为1/18000活产儿。在全球范围内,报告的发病率在每10万活产儿5例至32例之间,在亚太地区最高。女性受影响的频率略高于男性。常见的组织病理学表现是肝内和肝外胆管的炎性损伤,伴有硬化以及胆管树变窄甚至闭塞。若不治疗,这种情况会在生命的头几年导致肝硬化和死亡。BA并非遗传性疾病。对于BA的治疗,尚无有效的主要药物治疗方法。一旦怀疑患有BA,应尽快进行手术干预(Kasai肝门空肠吻合术),因为早期进行手术更有可能成功。如果Kasai手术未能恢复胆汁流动或出现肝硬化并发症,后期可能需要进行肝移植。目前,约90%的BA患者存活,且大多数患者生活质量正常。